
Our Health Library information does not replace the advice of a doctor. Please be advised that this information is made available to assist our patients to learn more about their health. Our providers may not see and/or treat all topics found herein.
Childhood Thyroid Cancer Treatment (PDQ®): Treatment - Health Professional Information [NCI]
General Information About Childhood Thyroid Cancer
Incidence
In the United States, the annual incidence of thyroid cancers is 10.7 cases per 1 million in people aged 0 to 19 years. The incidence is higher in females than in males (17.6 vs. 4.1 cases per 1 million people, respectively) and lower in Black people than in White people (3.9 vs. 11.8 cases per 1 million people, respectively). It accounts for approximately 6% of all cancers in this age group.[1] Thyroid cancer incidence is higher in children aged 15 to 19 years (34.4 cases per 1 million people), and it accounts for approximately 14% of all cancers arising in this older age group.[1] The trend toward larger tumors suggests that diagnostic scrutiny is not the only explanation for the observed results.[2]
Two time-trend studies using the Surveillance, Epidemiology, and End Results (SEER) Program database have shown a 2% and 3.8% annual increase in the incidence of differentiated thyroid carcinoma in the United States among children, adolescents, and young adults in the 1973 to 2011 and 1984 to 2010 periods, respectively.[2,3] Newer data from the National Childhood Cancer Registry show an average annual increase in incidence rates of 1.2% between 2012 and 2021, without changes in survival.[1] A similar trend has been documented in other countries.[4,5]
The papillary subtype is the most common subtype of childhood thyroid cancer, accounting for approximately 60% of cases, followed by the papillary follicular variant subtype (20%–25%), the follicular subtype (10%), and the medullary subtype (<10%). The anaplastic subtype occurs in less than 1% of pediatric thyroid carcinomas. The incidence of the papillary subtype and its follicular variant peaks between the ages of 15 and 19 years. The incidence of medullary thyroid cancer is the highest in children aged 0 to 4 years and declines at older ages (see Figure 1).[6]
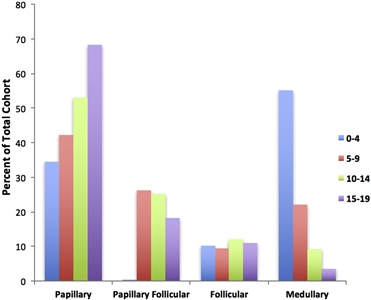
Figure 1. Incidence of pediatric thyroid carcinoma based on most frequent subtype per 100,000 as a percent of total cohort. Reprinted from International Journal of Pediatric Otorhinolaryngology, Volume 89, Sarah Dermody, Andrew Walls, Earl H. Harley Jr., Pediatric thyroid cancer: An update from the SEER database 2007–2012, Pages 121–126, Copyright (2016), with permission from Elsevier.
Diagnostic Evaluation
The prevalence of benign thyroid nodules in childhood has been estimated at about 0.5% to 2%.[7] However, thyroid nodules in children have a higher risk of malignancy (22%–26%) than thyroid nodules in adults (5%–15%).[8] Initial evaluation of a child or adolescent with a thyroid nodule includes the following:
- Ultrasonography of the thyroid and neck. Common ultrasonographic features of malignancy include hypoechogenicity, invasive margins, increased intranodular blood flow, microcalcifications, and abnormal cervical lymph nodes. Based on ultrasonographic characteristics, scoring systems have been developed to facilitate selection of nodules that require fine-needle aspiration (FNA) in adults. The most popular of these scoring systems is the Thyroid Imaging Reporting and Data System. However, the higher incidence of differentiated thyroid carcinoma in pediatric thyroid nodules and the lack of validation in the pediatric population limits the extrapolation of these criteria to children.[7,8]
- Serum thyroid-stimulating hormone (TSH) level. Thyroid function is usually normal. Hyperfunctioning nodules have a very low risk of malignancy (2%–6%).[8]
- Serum thyroglobulin level, which is usually elevated in differentiated thyroid carcinoma.
- FNA. The sensitivity, specificity, and accuracy of FNA in children are similar to those in adults, but there is a greater risk of false-negative findings in nodules larger than 4 cm.[8]
FNA results are categorized according to the six tiers of The Bethesda System for Reporting Thyroid Cytopathology (see Table 1).[8]
Table 1. Bethesda System for Reporting Thyroid Cytopathologya Bethesda Category Cytopathological Category Malignancy Rate Suggested Treatment FNA = fine-needle aspiration; US = ultrasonography. a Reprinted fromJournal of Pediatric Surgery, Volume 55, Issue 11, Emily R. Christison-Lagay, Reto M. Baertschiger, Catherine Dinauer, Gary L. Francis, Marcus M. Malek, Timothy B Lautz, Jennifer H. Aldrink, Christa Grant, Daniel S. Rhee, Peter Ehrlich, Roshni Dasgupta, Shahab Abdessalam, Pediatric differentiated thyroid carcinoma: An update from the APSA Cancer Committee, Pages 2273–2283, Copyright (2020), with permission from Elsevier.[8] I Nondiagnostic/inadequate 1%–5% Repeat FNA (other options: continued US surveillance, lobectomy) II Benign 0%–10% Serial US if small, lobectomy if >4 cm III Atypia/follicular lesion of undetermined significance 0%–44% Molecular genetics, lobectomy if no concerning mutation, thyroidectomy ifBRAFor fusion mutation IV Follicular neoplasm 60%–71% Molecular genetics, lobectomy if no concerning mutation, thyroidectomy ifBRAFor fusion mutation V Suspicious for malignancy 70%–86% Total thyroidectomy +/− central neck dissection VI Malignant 97%–100% Total thyroidectomy +/− central neck dissection While molecular testing of thyroid nodules could be helpful in the diagnosis of papillary thyroid carcinoma, there is no evidence to support its use.[7]
- Lymph node evaluation. Examination of the cervical lymph nodes is critically important in stratifying risk and determining operative strategies. Architecturally concerning features found on ultrasound in adults include round shape, irregular margins, calcifications, cystic change, peripheral vascularity, loss of fatty hilum, and heterogeneous echotexture. FNA should be performed on any suspicious lymph nodes in the lateral neck as confirmation of metastatic involvement before lateral neck dissection.[8]
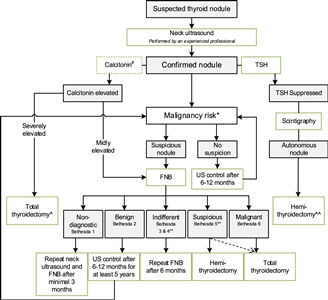
Figure 2. Flowchart showing the initial evaluation, treatment, and follow-up of pediatric thyroid nodules. #The expert panel suggests considering the measurement of serum calcitonin in children suspect of medullary thyroid carcinoma (MTC) based on individual conditions and the preference of the physician (Recommendation 5A). The expert panel suggests that, in selected cases (conditions that suggest MEN2, a positive family history of MEN2, or in case of bulky thyroid disease), the measurement of calcitonin may be of additional value for early diagnosis of MTC (Recommendation 5B). *Malignancy risk (suspicious vs. no suspicion) is based on neck ultrasound characteristics (described in section B2. Risk of malignancy in a thyroid nodule during childhood), history of radiation, and signs of a pre-disposition syndrome. If there is a significant increase in nodule size or the ultrasound characteristics change over time, repeated fine-needle biopsy (FNB) should be performed. **Analysis of the presence of other oncogenic drivers and gene fusions (e.g., RET/PTC and NTRK fusions) may be considered in Bethesda 3, 4, or 5 due to increasing awareness that these are also associated with the presence of papillary thyroid carcinoma (PTC). In case a BRAF V600E mutation is found, the risk of the thyroid nodule being malignant is high but needs to be confirmed, for example, by frozen section during thyroid surgery. ^Total thyroidectomy after proven presence of MTC. ^^Alternatively, FNB can be performed; in case of differentiated thyroid carcinoma (DTC), a total thyroidectomy should be performed. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. Credit: Lebbink, C. A., Links, T. P., Czarniecka, A., Dias, R. P., Elisei, R., Izatt, L., Krude, H., Lorenz, K., Luster, M., Newbold, K., Piccardo, A., Sobrinho-Simões, M., Takano, T., Paul van Trotsenburg, A. S., Verburg, F. A., & van Santen, H. M. (2022). 2022 European Thyroid Association Guidelines for the management of pediatric thyroid nodules and differentiated thyroid carcinoma. European Thyroid Journal, 11(6), e220146. Retrieved Aug 2, 2024, from https://doi.org/10.1530/ETJ-22-0146.
References:
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
- Vergamini LB, Frazier AL, Abrantes FL, et al.: Increase in the incidence of differentiated thyroid carcinoma in children, adolescents, and young adults: a population-based study. J Pediatr 164 (6): 1481-5, 2014.
- Golpanian S, Perez EA, Tashiro J, et al.: Pediatric papillary thyroid carcinoma: outcomes and survival predictors in 2504 surgical patients. Pediatr Surg Int 32 (3): 201-8, 2016.
- Pole JD, Zuk AM, Wasserman JD: Diagnostic and Treatment Patterns Among Children, Adolescents, and Young Adults with Thyroid Cancer in Ontario: 1992-2010. Thyroid 27 (8): 1025-1033, 2017.
- Schmidt Jensen J, Grønhøj C, Mirian C, et al.: Incidence and Survival of Thyroid Cancer in Children, Adolescents, and Young Adults in Denmark: A Nationwide Study from 1980 to 2014. Thyroid 28 (9): 1128-1133, 2018.
- Dermody S, Walls A, Harley EH: Pediatric thyroid cancer: An update from the SEER database 2007-2012. Int J Pediatr Otorhinolaryngol 89: 121-6, 2016.
- Lebbink CA, Links TP, Czarniecka A, et al.: 2022 European Thyroid Association Guidelines for the management of pediatric thyroid nodules and differentiated thyroid carcinoma. Eur Thyroid J 11 (6): , 2022.
- Christison-Lagay ER, Baertschiger RM, Dinauer C, et al.: Pediatric differentiated thyroid carcinoma: An update from the APSA Cancer Committee. J Pediatr Surg 55 (11): 2273-2283, 2020.
Differentiated Thyroid Cancer (Papillary / Follicular)
Risk Factors
Risk factors for pediatric differentiated thyroid cancer include the following:
- Radiation exposure. There is an excessive frequency of papillary thyroid adenoma and carcinoma after radiation exposure, as a result of either environmental contamination or use of ionizing radiation for diagnosis or treatment.[1,2,3,4] The risk increases after exposure to a mean dose of more than 0.05 Gy to 0.1 Gy (50–100 mGy), follows a linear dose-response pattern up to 30 Gy, and then declines. The risk of thyroid cancer after radiation exposure is greater at a younger age of exposure and persists more than 45 years after exposure.[4,5] Childhood cancer survivors with subsequent differentiated thyroid carcinomas tend to have, on average, smaller tumors and, more often, bilateral disease. However, no differences between survivors and controls have been documented in the occurrence of surgical complications, recurrence rate, or disease-related death.[6] For more information, see the Subsequent Neoplasms section in Late Effects of Treatment for Childhood Cancer.
Papillary thyroid carcinoma is the most frequent form of thyroid carcinoma diagnosed after radiation exposure.[5] Molecular alterations, including intrachromosomal rearrangements, are frequently found; among them, RET rearrangements are the most common.[5]
- Thyroid nodule and autoimmune thyroiditis. In a study of 485 nodules in 385 children who underwent fine-needle aspiration, thyroid cancer was present in 108 nodules (24%). Autoimmune thyroiditis, present in 95 patients (25%), was independently associated with an increased risk of thyroid cancer (odds ratio [OR], 2.19; 95% confidence interval [CI], 1.32–3.62). Papillary thyroid carcinoma was more common than follicular thyroid carcinoma. Among the papillary thyroid carcinomas, autoimmune thyroiditis was strongly associated with the diffuse sclerosing variant (OR, 4.74; 95% CI, 1.33–16.9).[7]
- Genetic factors. Genetic factors play a role in a subset of thyroid carcinomas. For thyroid carcinomas of follicular cells, only 5% to 10% are familial cancers. Of those, most familial cases are nonsyndromic, while a minority occur in the setting of well-defined cancer syndromes with known germline alterations, including the following:[8,9]
- APC-associated polyposis.
- Carney complex.
- PTEN hamartoma tumor syndrome.
- Werner syndrome.
- DICER1 syndrome.
Clinical Presentation and Prognostic Factors
Patients with thyroid cancer usually present with a thyroid mass with or without painless cervical adenopathy.[10] Based on medical and family history and clinical findings, the thyroid cancer may be part of a tumor predisposition syndrome such as APC-associated polyposis, PTEN hamartoma tumor syndrome, Carney complex, Werner syndrome, or DICER1 syndrome.[8,9]
In well-differentiated thyroid cancer, male sex, larger tumor size, and distant metastases have been found to have prognostic significance for early mortality. However, even patients in the highest risk group who had distant metastases had a 90% survival rate.[11]
In addition, the following observations have been reported:
- In a cross-sectional study involving 20% of community hospitals in the United States, the clinical presentation of 644 pediatric cases was compared with that of more than 43,000 adult cases. Compared with adults, children had a higher proportion of nodal involvement (31.5% in children vs. 14.7% in adults) and lung metastases (5.7% in children vs. 2.2% in adults).[10]
- Younger age is associated with a more aggressive clinical presentation in differentiated thyroid carcinoma. Higher recurrence rates have been associated with younger age at presentation.[12]
- Larger tumor size (>1 cm), extrathyroidal extension, and multifocal disease were associated with increased risk of nodal metastases.[13]
- Compared with pubertal adolescents, prepubertal children have a more aggressive presentation with a greater degree of extrathyroid extension, lymph node involvement, and lung metastases. However, outcomes are similar in the prepubertal and adolescent groups.[14,15,16]
- A French registry analysis found similar outcomes in children and young adults who developed papillary thyroid carcinoma after previous radiation therapy, compared with children and young adults who developed spontaneous papillary thyroid carcinoma. However, patients with previous thyroid irradiation for benign disease presented with more invasive tumors and lymph node involvement.[17]
- Tumor gene fusions in RET, ALK, and NTRK have been associated with high-risk clinical features in retrospective studies.
- In one study of 106 pediatric patients, 80 had identifiable genomic alterations, including 31 with fusion oncogenes (21 with RET, 6 with ALK, and 4 with NTRK). Patients with fusion-positive tumors were younger (aged <10 years, 93%); had a higher proportion of large tumors (>2 cm), extrathyroid extension, and lymph node and lung metastases; and had a higher incidence of recurrent or persistent disease than patients with BRAF-altered tumors. Expression of SLC5A5 (which encodes the sodium-iodide symporter protein, an important determinant of iodine I 131 [131I] avidity) was decreased in children with fusion-positive papillary thyroid carcinomas and in two patients with 131I-refractory disease who harbored an NTRK and RET fusion gene, respectively. The administration of larotrectinib and selpercatinib produced tumor responses and restored radioactive iodine uptake, underscoring the importance of molecular testing in pediatric patients with papillary thyroid cancer.[18]
- In a second study, 131 pediatric patients were categorized into three groups: RAS-altered (HRAS, KRAS, or NRAS), BRAF-altered (BRAF V600E), and RET or NTRK gene fusions (RET, NTRK1, or NTRK3 fusions).[19] Patients with RET or NTRK gene fusions were significantly more likely to have advanced lymph node disease and distant metastasis and less likely to achieve remission at 1 year, compared with patients in the RAS-altered and BRAF-altered groups.
- A study reported the outcomes of 65 Chinese patients (aged <20 years) with papillary thyroid carcinoma who presented with pulmonary metastases.[20] Twenty patients had persistent pulmonary metastases after treatment with radioiodine, designated as radioactive iodine–refractory (RAIR) disease. No significant difference in pathological characteristics was observed between patients younger than 15 years and patients aged 15 to 20 years, but younger patients were more likely to have RAIR disease (hazard ratio [HR], 3.500; 95% CI, 1.134–10.803; P = .023). RAIR disease was identified as an independent predictor of progressive disease (HR, 10.008; 95% CI, 2.427–41.268; P = .001). The Kaplan-Meier curve revealed lower progression-free survival (PFS) and disease-specific survival rates in the RAIR group than in the radioactive iodine–avid group (P < .001 and P = .039). Likewise, RAIR disease was a risk factor for unfavorable PFS in patients younger than 15 years (P < .001).
A review of the National Cancer Database found that patients aged 21 years and younger from lower-income families and those lacking insurance experienced a longer period from diagnosis to treatment of their well-differentiated thyroid cancer and presented with higher-stage disease.[21]
A single-institution retrospective review analyzed the impact of multifocal disease at presentation for patients with papillary thyroid carcinoma.[22] The study compared 283 children and adolescents with 5,564 adults. Multifocal disease was less common in children and adolescents with papillary thyroid carcinoma (45%; 127 of 283 patients) than in adults (54%; 3,023 of 5,564 adults; P = .002). There was no significant difference in 5-year recurrence-free probability, and the overall survival (OS) rate was 100% in both groups. There was no significant difference in the 5-year contralateral lobe papillary thyroid carcinoma–free probability between patients with unifocal disease and multifocal disease treated with lobectomy. The authors concluded that multifocal disease does not appear to warrant complete thyroidectomy in children and adolescents selected for lobectomy.
A single-institution study compared diagnostic whole-body 131I scans with stimulated thyroglobulin (sTg) levels as predictors of distant metastasis in children with papillary thyroid carcinoma.[23] A total of 142 patients (median age, 14.6 years; range, 4–18 years) were followed for 9.5 (±7.2) years and classified according to the American Thyroid Association risk of recurrence as low (28%), intermediate (16%), or high risk (56%). Of these patients, 127 had sTg evaluated. An sTg value of 21.7 ng/dL yielded a sensitivity of 88%, compared with 30% for diagnostic whole-body 131I scans, in predicting distant metastasis. Specificity was 60% for sTg levels and 100% for diagnostic whole-body 131I scans. Forty-two percent of patients obtained discordant results between diagnostic whole-body 131I scans and radioiodine therapy posttreatment whole-body 131I scans. In high-risk patients, sTg levels were particularly able to identify those who would have distant metastasis, with better diagnostic accuracy than whole-body 131I scans.
Histology and Molecular Features of Differentiated Thyroid Cancer
Tumors of the thyroid are classified as adenomas or carcinomas.[9,24] Adenomas are benign, well circumscribed, and encapsulated nodules that may cause notable enlargement of all or part of the gland, which extends to both sides of the neck. Some tumors may secrete hormones. Transformation to a malignant carcinoma may occur in some cells, which may grow and spread to lymph nodes in the neck or to the lungs. Approximately 20% of thyroid nodules in children are malignant.[9]
Histology
Papillary and follicular carcinomas are often referred to as differentiated thyroid carcinoma. The pathological classification of differentiated thyroid carcinomas is based on standard definitions set by the World Health Organization, and the criteria are the same for children and adults. Long-term outcomes for children and adolescents with differentiated thyroid carcinoma are excellent, with 10-year survival rates exceeding 95%.[9,25,26]
- Papillary thyroid carcinoma accounts for 90% or more of all cases of differentiated thyroid carcinoma occurring during childhood and adolescence. Pediatric papillary thyroid carcinoma may present with a variety of histological variants: classic, solid, follicular, and diffuse sclerosing.[27] Papillary thyroid carcinoma is frequently multifocal and bilateral, and it metastasizes to regional lymph nodes in most children. Hematogenous metastases to the lungs occur in up to 25% of cases.[9,28]
- Follicular thyroid carcinoma is uncommon. It is typically a unifocal tumor and more prone to initial hematogenous metastases to lungs and bones. Metastases to regional lymph nodes are uncommon. Histological variants of follicular thyroid cancer include Hürthle cell (oncocytic), clear cell, and insular (poorly differentiated) carcinoma.[9]
Molecular features
Thyroid tumorigenesis and progression of thyroid carcinomas of follicular cells (differentiated thyroid carcinoma, poorly differentiated papillary thyroid carcinoma, and anaplastic thyroid carcinoma) are defined by a multistep process that results in aberrant activation of the MAPK and/or PI3K/PTEN/AKT signaling pathways. Comprehensive genomic studies performed over the last decade have defined the landscape of these tumors, as well as their genotype-phenotype correlations. Using advanced sequencing technologies, oncogenic alterations are found in more than 90% of tumors.[29]
Variants in BRAF and RAS genes are the most common drivers, followed by gene fusions involving RET or NTRK:[8,30,31]
- BRAF: Single nucleotide variants of the BRAF gene are the most common alterations found in thyroid carcinoma. The most common variant is V600E (95% of BRAF-altered cases). BRAF variants are found in 40% to 80% of papillary thyroid carcinomas and in a lower proportion of poorly differentiated papillary thyroid carcinoma (5%–35%) and anaplastic thyroid carcinoma (10%–50%).[8,31]
The presence of BRAF V600E has been associated with extrathyroidal tumor extension and an increased risk of recurrence. However, its prognostic significance is controversial. BRAF V600E tumors appear to show a broadly immunosuppressive profile with high expression of anti–programmed death-ligand 1 (PD-L1).[8,31]
A retrospective analysis of 80 Brazilian patients younger than 18 years with papillary thyroid carcinoma identified AGK::BRAF fusions and BRAF V600E single nucleotide variants.[32]AGK::BRAF fusions, found in 19% of pediatric patients with papillary thyroid carcinoma, were associated with distant metastasis and younger age. BRAF V600E variants, found in 15% of patients with pediatric papillary thyroid carcinoma, were correlated with older age and larger tumor size.
- RAS: Oncogenic RAS activation can occur in any of the RAS family of genes (NRAS, HRAS, and KRAS), although the most frequent alterations are NRAS single nucleotide variants. RAS variants are markers of follicular-patterned thyroid lesions. They are present in 30% to 50% of follicular thyroid carcinomas, 25% to 45% of follicular variants of papillary thyroid carcinoma, and less than 10% of papillary thyroid carcinomas. They are also frequently found in poorly differentiated papillary thyroid carcinoma (20%–50%) and anaplastic thyroid carcinoma (10%–50%) and are believed to promote tumor progression. They have a higher prevalence in areas of iodine deficiency.[8,31]
- RET rearrangements: Multiple RET rearrangements have been identified in approximately 5% to 25% of papillary thyroid carcinomas and in less than 10% of its follicular variant. They are strongly associated with environmental or therapeutic radiation exposure. They are also common among young patients, many of whom present with nodal metastases and aggressive clinicopathological features.[8,31]RET variants have been reported to be more common in the diffuse sclerosing variant of papillary carcinoma than in standard nonsclerosing papillary carcinoma (83% vs. 15.4%; P = .0095).[27]
A retrospective review identified 113 RET fusion–positive tumors among 993 patients with papillary thyroid carcinoma.[33]RET fusion–positive tumors were three times more frequent in pediatric and adolescent patients (29.8%) than in adult patients (8.7%). A total of 20 types of RET fusions were identified. RET fusion–positive carcinomas were associated with aggressive tumor behavior, including high rates of lymph node metastases (75.2%) and distant metastases (18.6%). These rates were significantly higher than in carcinomas with NTRK fusions, BRAF V600E variants, and RAS variants. Local and distant metastases were also frequently found in patients with microcarcinomas positive for RET fusions. True recurrences occurred rarely (2.4%) and only in adult patients. The disease-specific survival rates were 99% at 2 years, 96% at 5 years, and 95% at 10 years.
- NTRK rearrangements: Rearrangements of NTRK1 and NTRK3 have been described in approximately 5% of papillary thyroid carcinomas. However, ETV6::NTRK3 fusion genes have been reported in 15% of radiation-induced papillary thyroid carcinomas. In young patients and children, NTRK-rearranged papillary thyroid carcinomas may present with lymph node metastases and aggressive clinicopathological features, similar to the presentation of RET-rearranged tumors.[8,31]
- DICER1 variants: Pathogenic variants of DICER1 have been identified in approximately 10% of papillary thyroid carcinomas.[34]DICER1 variants have also been described in a small cohort of patients with poorly differentiated thyroid carcinomas.[35]
A study correlated the status of hotspot DICER1 variants with clinical, histological, and outcome features in a series of 56 pediatric patients with papillary thyroid carcinomas. These patients had no clinical or family history of DICER1-related syndromic manifestations.[36] Fifteen papillary thyroid carcinomas (27%) harbored BRAF p.V600E. Eight cases of papillary thyroid carcinomas (14%) harbored DICER1 variants, with no associated BRAF p.V600E. DICER1 variants were identified in exons 26 and 27. A novel D1810del (c.5428_5430delGAT) variant was also detected. The study confirmed the absence of hotspot DICER1 variants in the matched nontumor tissue DNA in all eight DICER1-related papillary thyroid carcinomas. The study concluded that the increased incidence in female patients and enrichment in low-risk follicular-patterned papillary thyroid carcinomas are characteristics of DICER1-related papillary thyroid carcinomas.
A study profiled miRNA in 20 non-neoplastic thyroid tissue specimens, 8 adenomatous specimens, and 60 pediatric thyroid cancer specimens, 8 of which had DICER1 RNase IIIb variants. All differentiated thyroid cancers with DICER1 variants were follicular. Six were follicular variant papillary thyroid cancers, and two were follicular thyroid cancers.[37]
Other alterations include the following:[8,31]
- ALK rearrangements have been described in less than 10% of papillary thyroid carcinomas and are commonly associated with dedifferentiation.
- Activating variants of AKT1 have been described in 19% of recurrent or metastatic poorly differentiated papillary thyroid carcinomas.
- PPARG rearrangements are present in 20% to 50% of follicular thyroid carcinomas and in a lower proportion of follicular variants of papillary thyroid carcinoma.
- TERT-activating variants are commonly seen in poorly differentiated papillary thyroid carcinomas (20%–50%) and anaplastic thyroid carcinomas (30%–75%). These variants have also been reported in 10% to 35% of follicular thyroid carcinomas and 5% to 15% of papillary thyroid carcinomas. TERT variants are believed to promote tumor progression to poorly differentiated papillary thyroid carcinoma and anaplastic thyroid carcinoma and represent a negative prognostic marker.
- TP53 is altered in 40% to 80% of anaplastic thyroid carcinomas and 10% to 35% of poorly differentiated papillary thyroid carcinomas. It is considered a final step of tumor progression and a marker for poor prognosis.
The spectrum of somatic genetic alterations seems to differ between pediatric and adult patients when analyzing tumors with similar histologies, as follows:[29,30,38,39]
- Gene fusions involving RET or, less frequently, NTRK account for approximately 50% of the molecular alterations in pediatric differentiated thyroid carcinoma, compared with approximately 15% in adults.
- Gene alterations involving BRAF or RAS, which are present in approximately 70% of thyroid carcinomas diagnosed in adults, are noted in 20% to 40% of pediatric tumors. BRAF variants have been described in approximately 20% to 30% of cases, while RAS variants are much less frequently found in pediatrics (5%–10%).
- When combining evaluation of DNA and RNA, targetable alterations can be identified in approximately 98% of childhood thyroid carcinomas.
Treatment of Papillary and Follicular Thyroid Carcinoma
Treatment options for papillary and follicular (differentiated) thyroid carcinoma include the following:
Because differentiated thyroid cancer is rare in children, centralization of care to expert centers is highly encouraged.[6,9,24]
In 2015, the American Thyroid Association (ATA) Task Force on Pediatric Thyroid Cancer published guidelines for the management of thyroid nodules and differentiated thyroid cancer in children and adolescents. These guidelines are based on scientific evidence and expert panel opinion, with a careful assessment of the level of evidence.[9] In 2020 and 2022, the Cancer Committee of the American Pediatric Surgery Association and the European Thyroid Association (ETA) reviewed and expanded the ATA guidelines by incorporating more recent evidence.[24] The following sections of this summary provide an overview of the ATA guidelines and the proposed revisions, which are presented here without a specific endorsement by the National Cancer Institute (NCI).
Preoperative evaluation
Preoperative evaluation factors to consider include the following:
- Neck palpation and a comprehensive ultrasonography of all regions of the neck using a high-resolution probe and Doppler technique should be obtained by an experienced ultrasonographer. A complete ultrasonography examination should be performed before surgery.[6,9]
- The addition of cross-sectional imaging (contrast-enhanced computed tomography [CT] or magnetic resonance imaging) should be considered when there is concern about invasion of the aerodigestive tract. Importantly, if iodinated contrast agents are used, further evaluation and treatment with radioactive iodine may need to be delayed for 2 to 3 months until total body iodine burden decreases.[9]
- Chest imaging (x-ray or CT) may be considered for patients with substantial cervical lymph node disease.[9]
- Thyroid nuclear scintigraphy should be pursued only if the patient presents with suppressed thyroid-stimulating hormone (TSH).[9]
- The routine use of bone scan or fluorine F 18-fludeoxyglucose positron emission tomography (PET) is not recommended.[9]
- Further genetic or imaging diagnostics should be considered in cases of suspected familiar or extensive disease.[6]
Surgery
Total thyroidectomy is the cornerstone of the management of differentiated thyroid carcinoma. Pediatric thyroid surgery is ideally completed by a surgeon who has experience performing endocrine procedures in children and in a hospital with the full spectrum of pediatric specialty care. The ATA recommends that the thyroidectomy be performed by an experienced thyroid surgeon (>30 cases/year) or as a multidisciplinary approach between a pediatric surgeon and an adult endocrine or head and neck surgeon.[6,9]
Thyroidectomy
For patients with papillary or follicular carcinoma, total thyroidectomy is the recommended treatment. The ATA expert panel recommendation is based on data showing an increased incidence of bilateral (30%) and multifocal (65%) disease.[6,9]
In patients with a small unilateral tumor confined to the gland, a near-total thyroidectomy—in which a small amount of thyroid tissue (<1%–2%) is left in place at the entry point of the recurrent laryngeal nerve or superior parathyroid glands—might be considered to decrease permanent damage to those structures.[40]
A retrospective analysis identified factors associated with bilateral thyroid involvement in 115 pediatric patients with well-differentiated thyroid cancer.[41] Bilateral disease was present in 47 of 115 participants (41%). In multivariable analysis, only multifocality in the primary lobe was independently associated with bilateral disease (OR, 7.61; 95% CI, 2.44–23.8; P < .001). Among clinically node-negative patients with papillary carcinoma who did not have tumor multifocality in the primary lobe, bilateral disease was present in 5 of 32 patients (16%). The authors concluded that in children with differentiated thyroid cancer, tumor multifocality in the primary lobe is associated with bilateral disease, and they recommended prompt consideration of complete thyroidectomy after initial lobectomy.
Another multicenter retrospective analysis evaluated the prevalence of and risk factors for multifocal disease in 212 pediatric patients with papillary thyroid carcinoma.[42] The mean age at diagnosis was 14.1 years, and 23 patients were aged 10 years or younger. A total of 173 patients (82%) were female. Any amount of multifocal disease was present in 98 cases (46%), with bilateral multifocal disease present in 73 cases (34%). Predictors for multifocal and bilateral multifocal disease included age 10 years or younger, T3 tumor stage, and N1b nodal stage. The authors concluded that these risk factors and the high prevalence of multifocal disease should be considered when assessing the risks and benefits of surgical management options in pediatric patients with papillary thyroid carcinoma.
Thyroid resections that are less than a total thyroidectomy are associated with up to tenfold greater recurrence rates. Total thyroidectomy also optimizes the use of radioactive iodine for imaging and treatment.
Central neck dissection
A therapeutic central neck lymph node dissection (level VI nodes) should be done in the presence of clinical evidence of central or lateral neck metastases.[13]
For patients without clinical evidence of gross extrathyroidal invasion or locoregional metastasis, a prophylactic central neck dissection may be considered based on tumor focality and primary tumor size. However, because of the increased morbidity associated with central lymph node dissection, it is important to consider the risks and benefits of the extent of dissection on a case-by-case basis.[43]
Lateral neck dissection
Modified radical neck dissection is reserved for biopsy-proven metastatic disease in the lateral compartment (levels II, III, IV, and V). Cytological confirmation of metastatic disease to lymph nodes in the lateral neck is recommended before surgery.
Routine prophylactic lateral neck dissection is not recommended.
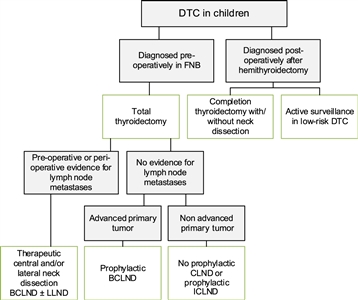
Figure 3. Flowchart showing the surgical approach for differentiated thyroid carcinoma (DTC) in children. BCLND, bilateral central lymph node dissection; CLND, central lymph node dissection; FNB, fine needle biopsy; ICLND, ipsilateral central lymph node dissection. ‘Active surveillance' in low-risk DTC implies ultrasound of the leftover thyroid tissue, including the evaluation of the cervical lymph nodes every 6–12 months by neck palpation and ultrasound. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. Credit: Lebbink, C. A., Links, T. P., Czarniecka, A., Dias, R. P., Elisei, R., Izatt, L., Krude, H., Lorenz, K., Luster, M., Newbold, K., Piccardo, A., Sobrinho-Simões, M., Takano, T., Paul van Trotsenburg, A. S., Verburg, F. A., & van Santen, H. M. (2022). 2022 European Thyroid Association Guidelines for the management of pediatric thyroid nodules and differentiated thyroid carcinoma. European Thyroid Journal, 11(6), e220146. Retrieved Aug 2, 2024, from https://doi.org/10.1530/ETJ-22-0146.
Classification and risk assignment
Despite the limited data in pediatrics, the ATA Task Force recommends the use of the tumor-node-metastasis (TNM) classification system to categorize patients into one of three risk groups.[9] This categorization strategy is meant to define the risk of persistent cervical disease and help determine which patients should undergo postoperative staging for the presence of distant metastasis.
- ATA pediatric low risk: Disease confined to the thyroid with N0 or NX disease or patients with incidental N1a (microscopic metastasis to a small number of central neck nodes). These patients are at lowest risk of distant disease but may still be at risk of residual cervical disease, especially if the initial surgery did not include central neck dissection.
- ATA pediatric intermediate risk: Extensive N1a or minimal N1b disease. These patients are at low risk of distant metastasis but are at an increased risk of incomplete lymph node resection and persistent cervical disease.
- ATA pediatric high risk: Regionally extensive disease (N1b) or locally invasive disease (T4), with or without distant metastasis. Patients in this group are at the highest risk of incomplete resection, persistent disease, and distant metastasis.
For more information about the TNM system, see the Stage Information for Thyroid Cancer section in Thyroid Cancer Treatment.
Postoperative staging and long-term surveillance
After surgical resection, disease is staged based on the operative findings to identify patients with persistent disease and those at intermediate or high risk of recurrence. Initial staging should be performed within 12 weeks after surgery to assess for evidence of persistent locoregional disease and to identify patients who are likely to benefit from additional therapy with 131I. The ATA pediatric risk level helps determine the extent of postoperative testing.[9] The standard imaging study for the follow-up of patients who have been treated for differentiated thyroid carcinoma is neck ultrasonography. It should be performed by a professional with experience using this procedure in children. The sensitivity and specificity of neck ultrasonography for recurrent differentiated thyroid carcinoma in follow-up for children who have been treated with total thyroidectomy are 85.7% and 89.4%, respectively.[6]
ATA pediatric low risk
- Initial postoperative staging includes a TSH-suppressed thyroglobulin. A diagnostic iodine I 123 (123I) scan is not required.
- TSH suppression should be targeted to serum levels of 0.5 to 1.0 mIU/L.
- In patients with no evidence of disease, surveillance should include ultrasonography at 6 months postoperatively and then annually for 5 years, as well as TSH-suppressed thyroglobulin levels every 3 to 6 months for 2 years and then annually.
- In children with positive thyroglobulin antibodies (common in patients with Hashimoto thyroiditis), trending thyroglobulin is less reliable, and a diagnostic 123I scan may be required.
ATA pediatric intermediate risk
- Initial postoperative staging includes a TSH-stimulated thyroglobulin and diagnostic 123I whole-body scan for further stratification and determination with 131I.
- TSH suppression should be targeted to serum levels of 0.1 to 0.5 mIU/L.
- In patients with no evidence of disease, surveillance should include ultrasonography at 6 months postoperatively and then every 6 to 12 months for 5 years (and then less frequently), as well as thyroglobulin levels (on hormone replacement therapy) every 3 to 6 months for 3 years and then annually.
- TSH-stimulated thyroglobulin and diagnostic 123I scan should be considered in 1 to 2 years for patients treated with 131I.
ATA pediatric high risk
- Initial postoperative staging includes a TSH-stimulated thyroglobulin and diagnostic 123I whole-body scan for further stratification and determination with 131I.
- TSH suppression should be targeted to serum levels of less than 0.1 mIU/L.
- In patients with no evidence of disease, surveillance should include ultrasonography at 6 months postoperatively and then every 6 to 12 months for 5 years (and then less frequently), as well as thyroglobulin levels (on hormone replacement therapy) every 3 to 6 months for 3 years and then annually.
- TSH-stimulated thyroglobulin and, possibly, a diagnostic 123I scan in 1 to 2 years in patients treated with 131I.
For patients with antithyroglobulin antibodies, deferred postoperative staging to allow time for antibody clearance, except in patients with T4 or M1 disease.
Radioactive iodine ablation (RAI)
The goal of 131I therapy is to decrease recurrence and mortality by eliminating iodine-avid disease.[6,9]
- The ATA Task Force recommends the use of 131I for the treatment of iodine-avid, persistent locoregional, or nodal disease that cannot be resected, and for known or presumed iodine-avid distant metastases. For patients with persistent disease after administration of 131I, the decision to pursue additional 131I therapy should be individualized based on clinical data and previous response. For patients without lymph node or distant metastases, there is no evidence that 131I can improve survival or reduce recurrence rates.[6]
- To facilitate 131I uptake by residual iodine-avid disease, the TSH level should be above 30 mIU/L. This level can be achieved by withdrawing levothyroxine for at least 14 days. A low-iodine diet should also be followed for 2 weeks before therapy. RAI should be deferred for 2 to 3 months after exposure to iodinated CT contrast, and urine iodine excretion should be confirmed to be less than 75 µ/L. In patients who cannot mount an adequate TSH response or cannot tolerate profound hypothyroidism, recombinant human TSH may be used.
- Therapeutic 131I administration is commonly based on either empiric dosing or whole-body dosimetry. Based on the lack of data comparing empiric treatment and treatment informed by dosimetry, the ATA Task Force was unable to recommend one specific approach. However, because of the differences in body size and iodine clearance in children compared with adults, all activities of 131I should be calculated by experts with experience in dosing children.
- A posttreatment whole-body scan is recommended for all children 4 to 7 days after 131I therapy. The addition of single-photon emission CT with integrated conventional CT (SPECT/CT) may help to distinguish the anatomic location of focal uptake.
While rare, late effects of 131I treatment include salivary gland dysfunction, bone marrow suppression, pulmonary fibrosis, and second malignancies.[44]
- Because response to 131I may be observed up to 15 to 18 months after therapy, long intervals of at least 12 months are suggested before re-treatment.[6]
Evidence (RAI):
- In a multicenter study of children and adolescents with differentiated thyroid carcinoma, 285 consecutive patients were treated with total thyroidectomy and RAI according to the ATA guidelines.[45]
- 87% of the patients had no evidence of active disease at a median follow-up of 133 months.
- In a single-center study of ATA pediatric low-risk differentiated thyroid cancer diagnosed between 2010 and 2020, 95 patients underwent total thyroidectomy followed by 131I therapy in 53% of patients.[46]
- There was no statistical difference in remission rates between patients treated with or without 131I therapy at 1 year (70% vs. 68.9%, respectively; P = .089) or last clinical evaluation (82% vs. 75.6%; P = .534).
- Over the study period, use of 131I in the patient population declined steadily, as the 2015 ATA Pediatric Differentiated Thyroid Cancer Guidelines recommended withholding 131I therapy in patients with low-risk disease. Accordingly, patients who received 131I therapy had longer follow-up (median, 5.8 years) than those who did not receive 131I therapy (median, 3.6 years).
The ETA has proposed a simplified follow-up plan based on thyroglobulin levels and neck ultrasonography (see Figure 4).[6]
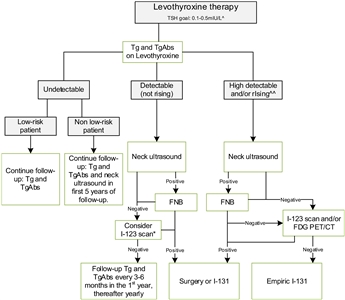
Figure 4. Flowchart showing the follow-up of children with differentiated thyroid carcinoma (DTC) who achieved complete remission after initial treatment with total thyroidectomy and I-131. This flowchart was developed for children with DTC who achieved complete remission defined as: undetectable levels of serum thyroglobulin (Tg) on levothyroxine (LT4), undetectable levels of Tg antibodies, negative neck ultrasound, and if performed, negative whole-body scan 1 year after last treatment. ^In the first year until clinical remission, TSH levels should be suppressed, while a normal low value of TSH (between 0.5 and 1.0 mIU/L) will be advisable thereafter. ^^The definition of consistently rising Tg on LT4 is debatable; the levels of Tg as well as the doubling time should be taken into account and weighted in the individual patient. *The expert panel suggests that, in children with detectable (but not rising) Tg and no focus on neck ultrasound, I-123 scanning may be considered in individual cases. When both ultrasound and radioiodine imaging did not yield a focus, FDG PET/CT may be considered. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. Credit: Lebbink, C. A., Links, T. P., Czarniecka, A., Dias, R. P., Elisei, R., Izatt, L., Krude, H., Lorenz, K., Luster, M., Newbold, K., Piccardo, A., Sobrinho-Simões, M., Takano, T., Paul van Trotsenburg, A. S., Verburg, F. A., & van Santen, H. M. (2022). 2022 European Thyroid Association Guidelines for the management of pediatric thyroid nodules and differentiated thyroid carcinoma. European Thyroid Journal, 11(6), e220146. Retrieved Aug 2, 2024, from https://doi.org/10.1530/ETJ-22-0146.
Treatment of Recurrent Papillary and Follicular Thyroid Carcinoma
Despite having more advanced disease at presentation than adults, children with differentiated thyroid cancer generally have an excellent survival with relatively few side effects.[25,47,48] For this reason, treatment of persistent or recurrent disease should be individualized, and the potential risks and benefits of therapy should be carefully considered. For children with persistent but not rising thyroglobulin levels on TSH suppression, primary neck ultrasonography is recommended; if negative, 123I scanning may be considered under TSH stimulation. If no residual or recurrent disease is found, serum thyroglobulin and serum thyroglobulin antibodies must be measured every 3 to 6 months. Patients with small cervical foci (i.e., <1 cm) or patients with cervical disease that cannot be visualized with cross-sectional imaging may be considered for (repeat) therapeutic 131I. However, these patients may also be safely observed while maintaining TSH suppression. Macroscopic cervical disease should be removed surgically if it can be safely accomplished. Children with pulmonary metastases may continue to experience posttherapy targeted 131I effects for years, and an undetectable thyroglobulin level should not be the focus of treatment efforts. As many as one-third of patients exhibit persistent but stable disease following RAI. Therapy should be considered only in patients who show signs of progression.[9,24]
Treatment options for recurrent papillary and follicular thyroid carcinoma include the following:
RAI with 131I
RAI with 131I is usually effective after recurrence.[49]
Tyrosine kinase inhibitors (TKIs)
For patients with 131I-refractory disease, molecularly targeted therapies using TKIs may provide alternative therapies.
TKIs with documented efficacy for the treatment of adults include the following:
- Sorafenib. Sorafenib is a VEGFR, PDGFR, and RAS kinase inhibitor. In a randomized phase III trial, sorafenib improved PFS when compared with placebo (10.8 months vs. 5.8 months) in adult patients with radioactive iodine–refractory locally advanced or metastatic differentiated thyroid cancer.[50] The U.S. Food and Drug Administration (FDA) approved sorafenib in 2013 for the treatment of adults with late-stage, metastatic differentiated thyroid carcinoma.
Pediatric-specific data are limited. However, in one case report, sorafenib produced a radiographic response in a patient aged 8 years with metastatic papillary thyroid carcinoma.[51]
- Lenvatinib. Lenvatinib is an oral VEGFR, FGFR, PDGFR, RET, and KIT inhibitor. In a phase III randomized study of adults with 131I-refractory differentiated thyroid cancer, lenvatinib was associated with a significant improvement in PFS and response rate when compared with a placebo.[52] The FDA approved lenvatinib in 2015 for the treatment of adults with progressive, radioactive iodine–refractory differentiated thyroid carcinoma.
Three children with papillary thyroid carcinoma who were refractory to radioactive iodine had a clinical response to lenvatinib.[53]
- Vemurafenib and dabrafenib (BRAF inhibitors). An open-label, nonrandomized, phase II study of vemurafenib was conducted in adult patients with papillary thyroid carcinoma that was 131I-refractory, metastatic or unresectable, and BRAF V600E variant positive. No participant had been previously treated with a TKI. A response rate of 38.5% was documented.[54] For patients with metastatic or advanced BRAF V600E–altered anaplastic thyroid carcinoma, the combination of dabrafenib with the MEK inhibitor trametinib showed a response rate of 69%.[55]
- Larotrectinib and entrectinib (NTRK inhibitors). Larotrectinib has been used to treat patients with TRK fusion–positive thyroid carcinoma. In one study, all five patients with TRK fusion–positive thyroid carcinomas who received larotrectinib therapy achieved partial or complete responses.[56] Responses to entrectinib have also been reported.[57] The FDA approved larotrectinib and entrectinib for the treatment of adults and children (restricted to patients older than 12 years for entrectinib) with solid tumors that include all of the following characteristics:[58]
- Have an NTRK gene fusion without a known acquired resistance variant.
- Are metastatic or for which surgical resection is likely to result in severe morbidity.
- Have no satisfactory alternative treatments or that have progressed following treatment.
- Selpercatinib (a RET inhibitor). In a phase I/II trial of selpercatinib therapy for patients (age range, 25–88 years) with RET-altered cancers, 19 patients with RET fusion–positive, previously treated thyroid cancers were enrolled.[59]
- Fifteen of 19 patients (79%) achieved objective responses (1 complete response and 14 partial responses), and the median duration of response was 18.4 months.
- The most common grades 3 to 4 treatment-related adverse events were hypertension (12%), increased alanine aminotransferase (10%) and aspartate aminotransferase (7%), diarrhea (3%), and prolonged QT interval (2%).
- In 2024, the FDA granted full approval to selpercatinib for the treatment of adult and pediatric patients aged 2 years and older with advanced or metastatic RET fusion–positive thyroid cancer who require systemic therapy and who are radioactive iodine–refractory (if radioactive iodine is appropriate).[60]
- Cabozantinib (a VEGFR and RET inhibitor). Cabozantinib and placebo were compared in a double-blind, phase III, randomized trial (COSMIC-311 [NCT03690388]) in adult patients (age range, 55–72 years). These patients had received at least one VEGFR-targeted TKI for differentiated thyroid carcinoma, and their disease was deemed progressive and radioactive iodine–refractory. An effective response was noted in 10 of 67 patients who received cabozantinib, compared with zero responses in the placebo group. The PFS was 11 months (95% CI, 7.4–13.8) in the cabozantinib arm, compared with 1.9 months (95% CI, 1.9–3.7) in the placebo arm, with an HR of 0.22 (95% CI, 0.14–0.31).[61] Based on these data, the FDA approved cabozantinib in this population.[62]
For more information, see Thyroid Cancer Treatment.
Treatment options under clinical evaluation for recurrent papillary and follicular thyroid carcinoma
Information about NCI-supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
References:
- Cahoon EK, Nadyrov EA, Polyanskaya ON, et al.: Risk of Thyroid Nodules in Residents of Belarus Exposed to Chernobyl Fallout as Children and Adolescents. J Clin Endocrinol Metab 102 (7): 2207-2217, 2017.
- Rose J, Wertheim BC, Guerrero MA: Radiation treatment of patients with primary pediatric malignancies: risk of developing thyroid cancer as a secondary malignancy. Am J Surg 204 (6): 881-6; discussion 886-7, 2012.
- Lal G, Groff M, Howe JR, et al.: Risk of subsequent primary thyroid cancer after another malignancy: latency trends in a population-based study. Ann Surg Oncol 19 (6): 1887-96, 2012.
- Lubin JH, Adams MJ, Shore R, et al.: Thyroid Cancer Following Childhood Low-Dose Radiation Exposure: A Pooled Analysis of Nine Cohorts. J Clin Endocrinol Metab 102 (7): 2575-2583, 2017.
- Iglesias ML, Schmidt A, Ghuzlan AA, et al.: Radiation exposure and thyroid cancer: a review. Arch Endocrinol Metab 61 (2): 180-187, 2017 Mar-Apr.
- Lebbink CA, Links TP, Czarniecka A, et al.: 2022 European Thyroid Association Guidelines for the management of pediatric thyroid nodules and differentiated thyroid carcinoma. Eur Thyroid J 11 (6): , 2022.
- Keefe G, Culbreath K, Cherella CE, et al.: Autoimmune Thyroiditis and Risk of Malignancy in Children with Thyroid Nodules. Thyroid 32 (9): 1109-1117, 2022.
- Acquaviva G, Visani M, Repaci A, et al.: Molecular pathology of thyroid tumours of follicular cells: a review of genetic alterations and their clinicopathological relevance. Histopathology 72 (1): 6-31, 2018.
- Francis GL, Waguespack SG, Bauer AJ, et al.: Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 25 (7): 716-59, 2015.
- Al-Qurayshi Z, Hauch A, Srivastav S, et al.: A National Perspective of the Risk, Presentation, and Outcomes of Pediatric Thyroid Cancer. JAMA Otolaryngol Head Neck Surg 142 (5): 472-8, 2016.
- Shayota BJ, Pawar SC, Chamberlain RS: MeSS: A novel prognostic scale specific for pediatric well-differentiated thyroid cancer: a population-based, SEER outcomes study. Surgery 154 (3): 429-35, 2013.
- Ye B, Shi J, Shen C, et al.: Comparison of differentiated thyroid carcinoma recurrence and its clinical features in children of different ages. Oncotarget 8 (29): 48051-48059, 2017.
- Kim J, Sun Z, Adam MA, et al.: Predictors of nodal metastasis in pediatric differentiated thyroid cancer. J Pediatr Surg 52 (1): 120-123, 2017.
- Lazar L, Lebenthal Y, Steinmetz A, et al.: Differentiated thyroid carcinoma in pediatric patients: comparison of presentation and course between pre-pubertal children and adolescents. J Pediatr 154 (5): 708-14, 2009.
- Redlich A, Luster M, Lorenz K, et al.: Age, American Thyroid Association Risk Group, and Response to Therapy Are Prognostic Factors in Children With Differentiated Thyroid Cancer. J Clin Endocrinol Metab 107 (1): e165-e177, 2022.
- Chesover AD, Vali R, Hemmati SH, et al.: Lung Metastasis in Children with Differentiated Thyroid Cancer: Factors Associated with Diagnosis and Outcomes of Therapy. Thyroid 31 (1): 50-60, 2021.
- Sassolas G, Hafdi-Nejjari Z, Casagranda L, et al.: Thyroid cancers in children, adolescents, and young adults with and without a history of childhood exposure to therapeutic radiation for other cancers. Thyroid 23 (7): 805-10, 2013.
- Lee YA, Lee H, Im SW, et al.: NTRK and RET fusion-directed therapy in pediatric thyroid cancer yields a tumor response and radioiodine uptake. J Clin Invest 131 (18): , 2021.
- Franco AT, Ricarte-Filho JC, Isaza A, et al.: Fusion Oncogenes Are Associated With Increased Metastatic Capacity and Persistent Disease in Pediatric Thyroid Cancers. J Clin Oncol 40 (10): 1081-1090, 2022.
- Tian T, Huang S, Dai H, et al.: Radioactive Iodine-Refractory Pulmonary Metastases of Papillary Thyroid Cancer in Children, Adolescents, and Young Adults. J Clin Endocrinol Metab 108 (2): 306-314, 2023.
- Garner EF, Maizlin II, Dellinger MB, et al.: Effects of socioeconomic status on children with well-differentiated thyroid cancer. Surgery 162 (3): 662-669, 2017.
- Scholfield DW, Lopez J, Eagan A, et al.: Is Multifocality a Predictor of Poor Outcome in Childhood and Adolescent Papillary Thyroid Carcinoma? J Clin Endocrinol Metab 108 (12): 3135-3144, 2023.
- Garcia Alves-Junior PA, de Andrade Barreto MC, de Andrade FA, et al.: Stimulated thyroglobulin and diagnostic 131-iodine whole-body scan as a predictor of distant metastasis and association with response to treatment in pediatric thyroid cancer patients. Endocrine 84 (3): 1081-1087, 2024.
- Christison-Lagay ER, Baertschiger RM, Dinauer C, et al.: Pediatric differentiated thyroid carcinoma: An update from the APSA Cancer Committee. J Pediatr Surg 55 (11): 2273-2283, 2020.
- Dermody S, Walls A, Harley EH: Pediatric thyroid cancer: An update from the SEER database 2007-2012. Int J Pediatr Otorhinolaryngol 89: 121-6, 2016.
- National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025.
- Brady C, Manning SC, Rudzinski E, et al.: Clinical Outcomes of Diffuse Sclerosing Variant Papillary Thyroid Carcinoma in Pediatric Patients. Laryngoscope 132 (5): 1132-1138, 2022.
- Sugino K, Nagahama M, Kitagawa W, et al.: Distant Metastasis in Pediatric and Adolescent Differentiated Thyroid Cancer: Clinical Outcomes and Risk Factor Analyses. J Clin Endocrinol Metab 105 (11): , 2020.
- Stosic A, Fuligni F, Anderson ND, et al.: Diverse Oncogenic Fusions and Distinct Gene Expression Patterns Define the Genomic Landscape of Pediatric Papillary Thyroid Carcinoma. Cancer Res 81 (22): 5625-5637, 2021.
- Bauer AJ: Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol Metab Clin North Am 46 (2): 389-403, 2017.
- Cancer Genome Atlas Research Network: Integrated genomic characterization of papillary thyroid carcinoma. Cell 159 (3): 676-90, 2014.
- Sisdelli L, Cordioli MICV, Vaisman F, et al.: AGK-BRAF is associated with distant metastasis and younger age in pediatric papillary thyroid carcinoma. Pediatr Blood Cancer 66 (7): e27707, 2019.
- Bulanova Pekova B, Sykorova V, Mastnikova K, et al.: RET fusion genes in pediatric and adult thyroid carcinomas: cohort characteristics and prognosis. Endocr Relat Cancer 30 (12): , 2023.
- Wasserman JD, Sabbaghian N, Fahiminiya S, et al.: DICER1 Mutations Are Frequent in Adolescent-Onset Papillary Thyroid Carcinoma. J Clin Endocrinol Metab 103 (5): 2009-2015, 2018.
- Chernock RD, Rivera B, Borrelli N, et al.: Poorly differentiated thyroid carcinoma of childhood and adolescence: a distinct entity characterized by DICER1 mutations. Mod Pathol 33 (7): 1264-1274, 2020.
- Onder S, Mete O, Yilmaz I, et al.: DICER1 Mutations Occur in More Than One-Third of Follicular-Patterned Pediatric Papillary Thyroid Carcinomas and Correlate with a Low-Risk Disease and Female Gender Predilection. Endocr Pathol 33 (4): 437-445, 2022.
- Ricarte-Filho JC, Casado-Medrano V, Reichenberger E, et al.: DICER1 RNase IIIb domain mutations trigger widespread miRNA dysregulation and MAPK activation in pediatric thyroid cancer. Front Endocrinol (Lausanne) 14: 1083382, 2023.
- Potter SL, Reuther J, Chandramohan R, et al.: Integrated DNA and RNA sequencing reveals targetable alterations in metastatic pediatric papillary thyroid carcinoma. Pediatr Blood Cancer 68 (1): e28741, 2021.
- Pekova B, Sykorova V, Dvorakova S, et al.: RET, NTRK, ALK, BRAF, and MET Fusions in a Large Cohort of Pediatric Papillary Thyroid Carcinomas. Thyroid 30 (12): 1771-1780, 2020.
- Spinelli C, Strambi S, Rossi L, et al.: Surgical management of papillary thyroid carcinoma in childhood and adolescence: an Italian multicenter study on 250 patients. J Endocrinol Invest 39 (9): 1055-9, 2016.
- Cherella CE, Richman DM, Liu E, et al.: Predictors of Bilateral Disease in Pediatric Differentiated Thyroid Cancer. J Clin Endocrinol Metab 106 (10): e4242-e4250, 2021.
- Banik GL, Shindo ML, Kraimer KL, et al.: Prevalence and Risk Factors for Multifocality in Pediatric Thyroid Cancer. JAMA Otolaryngol Head Neck Surg 147 (12): 1100-1106, 2021.
- Machens A, Elwerr M, Thanh PN, et al.: Impact of central node dissection on postoperative morbidity in pediatric patients with suspected or proven thyroid cancer. Surgery 160 (2): 484-92, 2016.
- Albano D, Bertagna F, Panarotto MB, et al.: Early and late adverse effects of radioiodine for pediatric differentiated thyroid cancer. Pediatr Blood Cancer 64 (11): , 2017.
- Cistaro A, Quartuccio N, Garganese MC, et al.: Prognostic factors in children and adolescents with differentiated thyroid carcinoma treated with total thyroidectomy and RAI: a real-life multicentric study. Eur J Nucl Med Mol Imaging 49 (4): 1374-1385, 2022.
- Bojarsky M, Baran JA, Halada S, et al.: Outcomes of ATA Low-Risk Pediatric Thyroid Cancer Patients Not Treated With Radioactive Iodine Therapy. J Clin Endocrinol Metab 108 (12): 3338-3344, 2023.
- Golpanian S, Perez EA, Tashiro J, et al.: Pediatric papillary thyroid carcinoma: outcomes and survival predictors in 2504 surgical patients. Pediatr Surg Int 32 (3): 201-8, 2016.
- Vergamini LB, Frazier AL, Abrantes FL, et al.: Increase in the incidence of differentiated thyroid carcinoma in children, adolescents, and young adults: a population-based study. J Pediatr 164 (6): 1481-5, 2014.
- Powers PA, Dinauer CA, Tuttle RM, et al.: Treatment of recurrent papillary thyroid carcinoma in children and adolescents. J Pediatr Endocrinol Metab 16 (7): 1033-40, 2003.
- Brose MS, Nutting CM, Jarzab B, et al.: Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 384 (9940): 319-28, 2014.
- Iyer P, Mayer JL, Ewig JM: Response to sorafenib in a pediatric patient with papillary thyroid carcinoma with diffuse nodular pulmonary disease requiring mechanical ventilation. Thyroid 24 (1): 169-74, 2014.
- Schlumberger M, Tahara M, Wirth LJ, et al.: Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 372 (7): 621-30, 2015.
- Mahajan P, Dawrant J, Kheradpour A, et al.: Response to Lenvatinib in Children with Papillary Thyroid Carcinoma. Thyroid 28 (11): 1450-1454, 2018.
- Brose MS, Cabanillas ME, Cohen EE, et al.: Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol 17 (9): 1272-82, 2016.
- Subbiah V, Kreitman RJ, Wainberg ZA, et al.: Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer. J Clin Oncol 36 (1): 7-13, 2018.
- Drilon A, Laetsch TW, Kummar S, et al.: Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med 378 (8): 731-739, 2018.
- Chu YH, Dias-Santagata D, Farahani AA, et al.: Clinicopathologic and molecular characterization of NTRK-rearranged thyroid carcinoma (NRTC). Mod Pathol 33 (11): 2186-2197, 2020.
- Bayer HealthCare Pharmaceuticals: VITRAKVI (larotrectinib): Prescribing Information. Stamford, Conn: Loxo Oncology, Inc., 2018. Available online. Last accessed November 29, 2024.
- Wirth LJ, Sherman E, Robinson B, et al.: Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N Engl J Med 383 (9): 825-835, 2020.
- Eli Lilly and Company: RETEVMO (selpercatinib): Prescribing Information. Indianapolis, Ind: Lilly USA, LLC, 2024. Available online. Last accessed November 29, 2024.
- Brose MS, Robinson B, Sherman SI, et al.: Cabozantinib for radioiodine-refractory differentiated thyroid cancer (COSMIC-311): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 22 (8): 1126-1138, 2021.
- Duke ES, Barone AK, Chatterjee S, et al.: FDA Approval Summary: Cabozantinib for Differentiated Thyroid Cancer. Clin Cancer Res 28 (19): 4173-4177, 2022.


This information does not replace the advice of a doctor. Ignite Healthwise, LLC disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use and Privacy Policy. Learn how we develop our content.
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.