
Our Health Library information does not replace the advice of a doctor. Please be advised that this information is made available to assist our patients to learn more about their health. Our providers may not see and/or treat all topics found herein.
Childhood Vascular Tumors Treatment (PDQ®): Treatment - Health Professional Information [NCI]
Benign Tumors
Benign vascular tumors include the following:
- Infantile hemangioma.
- Congenital hemangioma.
- Hepatic vascular tumors.
- Spindle cell hemangioma.
- Epithelioid hemangioma.
- Pyogenic granuloma (lobular capillary hemangioma).
- Angiofibroma.
- Juvenile nasopharyngeal angiofibroma.
Juvenile nasopharyngeal angiofibroma is not included in the World Health Organization (WHO) classification or the International Society for the Study of Vascular Anomalies (ISSVA) classification of vascular tumors. It is included here because growing evidence reveals vascular differentiation and proliferation in these tumors with response to vascular remodeling and antiproliferative agents.
Infantile Hemangioma
Incidence and epidemiology
Infantile hemangiomas (IH) are the most common benign vascular tumor of infancy, occurring in 4% to 5% of infants. The true incidence is unknown.[1] They are not usually present at birth and are diagnosed most commonly at age 3 to 6 weeks.[2,3,4,5] The lesion proliferates for an average of 5 months, stabilizes, and then involutes over several years.
Infantile hemangiomas are more common in females, non-Hispanic White patients, and premature infants. Multiple hemangiomas are more common in infants who are the product of multiple gestations or in vitro fertilization.[5,6,7] Infantile hemangiomas are associated with advanced maternal age, placenta previa, pre-eclampsia, and other placental anomalies.[5]
Clinical presentation
Most infantile hemangiomas are not present at birth, but precursor lesions such as telangiectasia or faint discoloration of the skin or hypopigmentation can often be seen. The lesion can be mistaken as a bruise from birth trauma or as a capillary malformation (port-wine stain) (see Figure 1).[8,9]
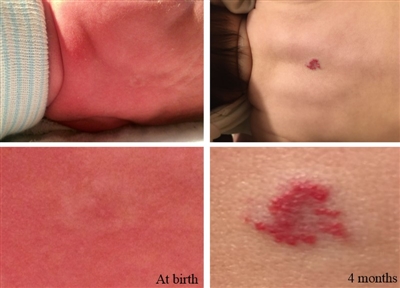
Figure 1. The photos on the left depict the precursor lesion (faint color with halo). The photos on the right depict the hemangioma after proliferation (slightly raised with a brighter central color). Credit: Israel Fernandez-Pineda, M.D.
Infantile hemangiomas can be superficial in the dermis, deep in the subcutaneous tissue, combined, or in the viscera. Combined lesions are common and generally appear in the head and neck but can be anywhere on the body.
Infantile hemangiomas can be characterized as follows:
- Local: Most lesions are localized and noted to be in a well-defined area without evidence of a geometric pattern.
- Segmental: Most segmental hemangiomas occur in the head and neck region (PHACE syndrome) but can be seen in the genitourinary area, arm, chest, or legs (PELVIS/LUMBAR/SACRAL syndrome).
- Diffuse hemangiomas of the face demonstrate defined cutaneous patterns. Several studies have evaluated the distributions of these hemangiomas and found the following four distinct patterns or segments:
- Segment 1 involves the lateral forehead, anterior temporal scalp, and the lateral frontal scalp.
- Segments 2 and 3 are located over the maxillary and mandibular area.
- Segment 4 covers the medial frontal scalp, nose, and philtrum.
Two papers have noted this observation and suggest the involvement of neural crest derivatives in facial hemangioma development.[10,11] Segmental hemangiomas commonly occur in females and are more likely associated with complications and other syndromes.[12,13]
For information about PHACE syndrome or PELVIS/LUMBAR/SACRAL syndrome, see the Syndromes associated with infantile hemangioma section.
- Diffuse hemangiomas of the face demonstrate defined cutaneous patterns. Several studies have evaluated the distributions of these hemangiomas and found the following four distinct patterns or segments:
- Multiple: More than one lesion but noted in the past as greater than five lesions, because of the increased risk of visceral involvement (mostly the liver).
The cutaneous appearance of infantile hemangiomas is usually red to crimson, firm, and warm in the proliferative phase. The lesion then lightens centrally and becomes less warm and softer; it then flattens and loses its color. The process of involution can take several years and once involution has occurred, regrowth is uncommon. In two patients treated with growth hormone, regrowth after involution was noted.[14] On further investigation, growth hormone receptors were found on the infantile hemangioma cells. Although preliminary, this may advance the research into the etiology of hemangioma growth.
Ulceration is the most common complication of infantile hemangiomas, occurring in 10% to 15% of patients. Ulceration typically occurs during the proliferative phase, and it can lead to bleeding and secondary infections.[15] Most other complications in the proliferative phase result from the impact of the mass on local structures (e.g., visual or auditory compromise, airway obstruction).[16]
Permanent sequelae, such as telangiectasia, anetodermal skin, redundant skin, and a persistent superficial component, can occur after hemangioma involution. Hemangiomas with a history of ulceration are more likely to cause scarring and potential local anatomical complications.[15] Rare instances of dysesthesias in sites of involuted infantile hemangiomas in the absence of ulceration have been described.[17][Level of evidence C1] In a retrospective cohort study of 184 hemangiomas, the overall incidence of significant sequelae was 54.9%. Sequelae were more common in combined hemangiomas, hemangiomas with a step or abrupt border, and cobblestone surface hemangiomas. Furthermore, this study revealed that the average age to hemangioma involution was 3.5 years.[18] Thus, prophylactic measures such as maintaining dermal integrity with moisturizing barrier agents are indicated for infantile hemangiomas and are important before and during the proliferative phase.[16] Once an ulceration has occurred, it is important to aggressively manage the ulceration to promote healing, prevent infection, and treat pain. In addition to pain control, management includes steroid ointments, antibiotic ointments or systemic antibiotics, laser therapy, or topical timolol.[16]
Biology and histopathology
Most infantile hemangiomas occur sporadically. However, they may rarely be caused by an abnormality of chromosome 5 and present in an autosomal dominant pattern.[19] In a study that evaluated inheritance patterns of infantile hemangiomas, 34% of patients had a family history of infantile hemangioma, most commonly in a first-degree relative.[19,20]
The exact mechanism that causes the initial proliferation of blood vessels followed by involution of the vascular component of hemangioma and replacement of fibrofatty tissue is unknown. Several cell types have been isolated from hemangiomas: progenitor/stem cells (HemSC), endothelial cells (HemEC), pericytes (HemPericytes), and mast cells.[21,22] These cells appear to play a role in the development of infantile hemangiomas.
HemSC represent a small percentage of proliferating hemangioma cells and have the ability for self renewal and multilineage differentiation. These cells differentiate into endothelial cells, adipocytes, and pericytes. When HemSC are implanted into immunodeficient mice, hemangioma-like lesions form and then spontaneously regress, similar to infantile hemangiomas.[23] This suggests that infantile hemangioma proliferation occurs during vasculogenesis (the formation of new blood vessels from angioblasts), as opposed to angiogenesis (the formation of new blood vessels from existing blood vessels).
HemEC are plump, metabolically active, and resemble fetal endothelial cells in the proliferative phase. Evaluation of infantile hemangioma endothelial cells suggest that they are clonal in nature.[23,24,25]
HemPericytes surround the vasculature and are abundant in the proliferative phase. These cells express markers of pericytes and smooth muscle cells, such as neural-glial antigen 2 (NG2), platelet-derived growth factor receptor beta (PDGFR-beta), calponin, alpha smooth muscle actin (SMA), and NOTCH3. HemPericytes are proangiogenic, as they express increased vascular endothelial growth factor A (VEGF-A), decreased angiopoietin-1 (ANGPT1), increased proliferation, increased vessel formation in vivo, and decreased ability to suppress proliferation.[26] One study reported that proliferating infantile hemangiomas contained higher levels of messenger RNA, proteins for NOTCH1, 3, and 4 receptors and their ligands, and the downstream coactivator MAML1 than did normal skin, involuting infantile hemangiomas, and propranolol-treated infantile hemangiomas.[27]
Mast cells are found largely in the early involuting phase, but they are also found in small numbers in the proliferative phase and at the end of involution. Their function in infantile hemangiomas is unknown but they have been shown to play a role in other skin tumors such as basal cell carcinoma, squamous cell carcinoma, and melanoma.[22]
Provasculogenic factors are expressed during proliferation; these factors include VEGF, fibroblast growth factor (FGF), CD34, CD31, CD133, lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1), and insulin-like growth factor 2 (IGF-2).[28,29,30,31] During involution, infantile hemangiomas show increased apoptosis.[31] During this phase, there are also increased mast cells and levels of metalloproteinase, as well as upregulation of interferon and decreased basic FGF (bFGF).[31,32,33] Throughout proliferation and involution, endothelial cells in infantile hemangioma express a particular phenotype showing positive staining for GLUT1 and placenta-associated antigens (Fc-gamma receptor II, merosin, and Lewis Y antigen). These markers are absent in normal capillaries and in other vascular tumors such as congenital hemangioma and vascular malformations. Placental chorionic villi share these same markers. However, no relationship between hemangiomas and placental chorionic villi has been found.[28]
Hypoxia appears to have a critical role in the pathogenesis of hemangiomas. There is an association between hemangiomas and placental hypoxia, which is increased in prematurity, multiple pregnancies, and placental anomalies.[2,5] Multiple targets of hypoxia [34,35] are demonstrated in proliferating hemangiomas, including VEGF-A, GLUT1, and IGF-2.[28,30,36] The hypothesis suggests that a proliferating hemangioma is an attempt to normalize hypoxic tissue that occurred in utero.
Diagnostic evaluation
Infantile hemangiomas are usually diagnosed by the history and clinical appearance. Biopsy is rarely needed and performed only if there is an atypical appearance and/or atypical history and presentation. Imaging is not usually necessary, but diagnostic ultrasonography is beneficial if there is a deeper lesion without a cutaneous component and reveals a well-circumscribed, hypoechoic, high-flow lesion with a typical Doppler wave characteristic.[37] Additionally, infants with five or more cutaneous hemangiomas should undergo ultrasonography of the liver to screen for hepatic hemangioma.[38]
Infantile hemangioma with minimal or arrested growth
Infantile hemangioma with minimal or arrested growth (IH-MAG) is a variant of hemangioma that can be confused with capillary malformation because of their unusual characteristics. These hemangiomas are mostly fully formed at birth and are characterized by telangiectasia and venules with light and dark areas of skin coloration (see Figure 2). They resolve spontaneously and are pathologically GLUT1 positive.[39] They are mainly located on the lower body but can be present in the head and neck area. If they are segmental, they can be associated with PHACE syndrome.[40] Associated soft tissue hypertrophy may persist through childhood.[41]
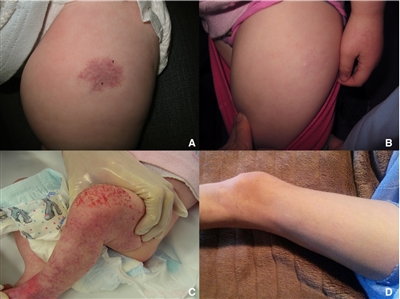
Figure 2. Patient 4 at (A) presentation and (B) resolution. Patient 5 at (C) presentation and (D) resolution. Ma, E. H., Robertson, S. J., Chow, C. W., and Bekhor, P. S. (2017), Infantile Hemangioma with Minimal or Arrested Growth: Further Observations on Clinical and Histopathologic Findings of this Unique but Underrecognized Entity. Pediatr Dermatol, 34: 64–71. doi:10.1111/pde.13022. Used with permission.
Airway infantile hemangioma
Airway infantile hemangiomas are usually associated with segmental hemangiomas in a bearded distribution, which may include all or some of the following—the preauricular skin, mandible, lower lip, chin, or anterior neck. It is important for an otolaryngologist to proactively assess lesions in this distribution before signs of stridor occur. Airway infantile hemangioma incidence increases with a larger area of bearded involvement.[42]
Airway infantile hemangiomas can occur without skin lesions. A retrospective study of the Vascular Anomaly Database at the Children's Hospital of Pittsburgh analyzed 761 cases of infantile hemangioma. Thirteen patients (1.7%) had subglottic hemangiomas. Of those 13 patients, 4 (30%) had bearded distributions, 2 (15%) had cutaneous hemangiomas, and 7 (55%) had no cutaneous lesions.[43] For information about the treatment of airway infantile hemangiomas, see the Propranolol therapy section.
Ophthalmologic involvement of hemangiomas
Periorbital hemangiomas can cause visual compromise.[44] This usually occurs with hemangiomas of the upper medial eyelid but any hemangioma around the eye that is large enough can distort the cornea or obstruct the visual axis. Subcutaneous periocular hemangiomas can extend into the orbit, causing exophthalmos or globe displacement with only limited cutaneous manifestations. Issues with these lesions include astigmatism from direct pressure of the growing hemangioma, ptosis, proptosis, and strabismus. One of the leading causes of preventable blindness in children is stimulus-deprivation amblyopia caused by hemangioma obstruction. All periorbital hemangiomas or those with any possibility of potential visual impairment should have an ophthalmologic evaluation.
Two institutions in France and Canada performed a retrospective analysis of patients in a vascular anomalies practice. The investigators reviewed the records of all patients with a diagnosis of segmental facial or periorbital focal infantile hemangioma who had clinical photographs and brain magnetic resonance imaging (MRI) available.[45][Level of evidence C1] The study included 122 children (90 girls, 32 boys; mean age, 16.6 months). Forty-five children (36.9%) had a facial infantile hemangioma larger than 5 cm. Twenty-two patients (18.0%) had PHACES or possible PHACES syndrome. Cerebrovascular structural anomalies were seen in 14 of 22 patients with PHACES syndrome and no patients without PHACES syndrome. Brain anomalies were seen in 6 of 22 patients with PHACES syndrome and 1 patient without PHACES syndrome (P < .001). Cardiovascular anomalies were seen in six patients, and ocular anomalies were seen in eight patients. Of these 14 patients, 13 had PHACES syndrome. The authors concluded that clinical concern about associated extracutaneous anomalies is warranted for all children with facial segmental or periorbital focal infantile hemangiomas, including those with small hemangiomas.
Infantile hemangiomas can occur in the conjunctiva (see Figure 3). These hemangiomas can be associated with other ophthalmologic abnormalities and are treated with oral or topical beta-blockers.[46]
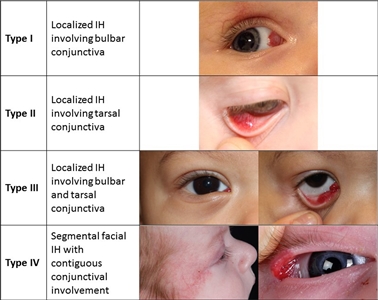
Figure 3. Proposed classification of infantile hemangiomas involving the conjunctiva. Theiler M, Baselga E, Gerth-Kahlert C, et al. Infantile hemangiomas with conjunctival involvement: An underreported occurrence. Pediatr Dermatol. 2017;34:681–685. https://doi.org/10.1111/pde.13305 Copyright © 2017 John Wiley & Sons, Inc.
Syndromes associated with infantile hemangioma
PHACE syndrome
Posterior fossa–brain malformations; Hemangiomas; Arterial, Cardiac, and Eye abnormalities (PHACE) syndrome: PHACE syndrome represents a spectrum of diseases and is defined by the presence of large segmental infantile hemangiomas, usually on the face or head, but can include the neck, chest, or arm, in association with one or more congenital malformations (see Figure 4).[47] PHACE syndrome is more common in girls and in full-term, normal birth weight and singleton infants.[13,48,49,50,51,52] The syndrome is not rare among patients with infantile hemangiomas. A prospective study of 108 infants with large facial hemangiomas observed that 31% of patients had PHACE syndrome.[53] Rare cases of PHACE syndrome have been reported in infants with hemangiomas smaller than 5 cm.[45][Level of evidence C1]
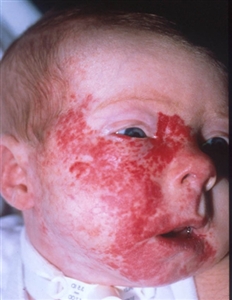
Figure 4. A large segmental infantile hemangioma (plaque-like) in a bearded distribution. This patient has an increased risk of PHACE syndrome, airway infantile hemangioma, and ulceration. A tracheostomy was placed secondary to a very diffuse airway hemangioma. Credit: Denise Adams, M.D. Garzon MC, Epstein LG, Heyer GL, et al.: PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr 178: 24-33.e2, 2016. PMID: 27659028
Consensus criteria for definite and possible PHACE syndrome were updated at an expert panel meeting, as follows:[47]
PHACE
- Posterior fossa abnormalities. Posterior fossa malformations include Dandy-Walker complex, cerebellar hypoplasia, atrophy, and dysgenesis/agenesis of the vermis. Effects of these anomalies include developmental delays and pituitary dysfunction.[54]
- Hemangiomas.[53,55,56,57,58]
- A large segmental hemangioma over the face and/or scalp with a surface area of 22 cm2 or greater (5 cm × 4.5 cm).
- A large segmental hemangioma of the neck, trunk, or proximal upper extremity.
Infants with two major criteria of PHACE (e.g., supraumbilical raphe and coarctation of the aorta) but lacking cutaneous infantile hemangiomas should undergo complete evaluation for PHACE.
- Arterial abnormalities. Cerebrovascular anomalies can include carotid artery abnormalities (including tortuosity) and absence, dilation/aneurysm, or narrowing of cerebral vessels. These anomalies, especially related to the carotid arteries, can lead to progressive arterial occlusion and even stroke. The risk categories are as follows:[51,52,59,60,61]
- Low risk: This category includes patients with arterial anomalies frequently seen in a general screening population. It also includes findings that have either no or very minimal clinical impact on patient outcome, even if rarely seen in the general population. Examples are persistent embryonic arteries, anomalous arterial origin or course, and circle of Willis variants.
- Intermediate risk: Includes patients with nonstenotic dysgenesis, including those with ectatic or segmentally enlarged arteries. It also includes patients with a narrowing or occlusion of arteries proximal to the circle of Willis, with no perceived hemodynamic risk. An evaluation of the patency of the circle of Willis is essential.
- High risk: This category includes patients with one or more of the following:
- Significant narrowing (>25%) or occlusion of principal cerebral vessels within or above the circle of Willis that results in an isolated circulation.
- Tandem or multiple arterial stenoses associated with complex blood flow that may potentially result in diminished cerebral perfusion. Patients with cerebrovascular stenosis in the setting of coarctation of the aorta are likely at higher risk of transient and permanent neurological ischemic events.
- Imaging findings in the brain parenchyma suggestive of chronic or silent ischemia, or progressive steno-occlusive disease. These parenchymal brain MRI findings include existing infarction, chronic or border zone ischemic changes, and presence of lenticulostriate collateral dilation or pial collaterals.
- Cardiac abnormalities. Aortic arch anomalies observed in PHACE syndrome are unusually complex, with involvement of the transverse and descending aorta arch. The arch obstruction is most often long-segment. The obstruction is frequently characterized by areas of arch narrowing with adjacent segments of marked aneurysmal dilatation.
- Eye abnormalities. Ophthalmologic anomalies can include microphthalmos, retinal vascular abnormalities, persistent fetal retinal vessels, exophthalmos, coloboma, and optic nerve atrophy. These abnormalities are rare and occur in 7% to 10% of patients.[62]
A retrospective review identified midline rhabdomyomatous mesenchymal hamartomas and chin hamartomas in a small number of children with PHACE or LUMBAR syndrome.[63] These are not currently included as minor criteria.
Diagnosis of PHACE syndrome requires clinical examination, cardiac evaluation with echocardiogram, ophthalmologic evaluation, and MRI/magnetic resonance angiogram (MRA) of the head and neck. All patients with intermediate-risk and high-risk central nervous system (CNS) findings should be monitored by a neurologist and/or neurosurgeon. Coarctation of the aorta requires immediate cardiology consultation, and a cardiac MRI/MRA may be warranted. A report of two patients with retro-orbital infantile hemangiomas and arteriopathy suggested a possible new presentation of PHACE syndrome.[58] For patients with proptosis, globe deviation, and strabismus, an MRI/MRA is recommended. Further workup for PHACE syndrome may be needed based on CNS findings.
Short- and long-term issues related to PHACE syndrome include the following:[64,65,66]; [67][Level of evidence C1]
- Headache.
- May present at an early age.
- Can be severe.
- New-onset headaches should be evaluated for vasculopathy and/or cerebral ischemia.
- Neurology referral is recommended.
- Vasoconstrictive medications are contraindicated.
- Hearing loss and speech-language delays.
- Speech-language delays may be a consequence of hearing deficits, prolonged hospitalizations, or may occur because of other neurodevelopmental anomalies.
- Sensorineural hearing loss is the most common type, and it is usually ipsilateral to the infantile hemangioma, which may involve the ipsilateral cranial nerve VIII.
- Early detection is crucial.
- All patients with PHACE syndrome should undergo hearing screening as a newborn and at least one follow-up if initial screening is normal.
- Dysphagia, feeding disorders, speech disorders, and/or language delay.
- Increased in patients with posterior fossa malformations, lip/oropharynx or airway hemangiomas, hearing loss, and those with a history of cardiac surgery.
- Patients should undergo an initial speech language evaluation before age 24 months.
- Patients with feeding difficulties should be referred for evaluation by a pediatric speech-language pathologist at any age.
- Dysphagia may be secondary to the disease location (lip, oral cavity, and pharynx) or oral motor coordination.
- Endocrine abnormalities.
- Thyroid dysfunction and hypopituitarism resulting in growth hormone deficiency are the most frequently reported abnormalities.
- Other manifestations of hypopituitarism, including hypogonadotropic hypogonadism and adrenal insufficiency, have been described.
- Patients should undergo neonatal screening and repeat studies if symptomatic.
- Growth hormone deficiency: Most reported cases are associated with hypopituitarism with empty or partially empty sella turcica noted on MRI, but it may also occur without evidence of central nervous system malformations.
- Neonatal hypoglycemia can be a sign of hypopituitarism and should prompt additional endocrinologic evaluation.
- Other consequences of pituitary dysfunction include hypogonadotropic hypogonadism, manifesting with delayed pubertal onset and late-onset adrenal insufficiency. These findings emphasize the importance of focused assessment of height, weight, and developmental milestones in the care of children with PHACE.
- Dental abnormalities (enamel hypoplasia).
- A study of 18 children with PHACE or possible PHACE syndrome revealed that 28% of patients had enamel hypoplasia. All of the affected children had intraoral hemangiomas. Five of 11 (45%) patients with intraoral hemangiomas had enamel defects. Children with enamel hypoplasia are at increased risk of developing caries.
- Patients should be examined for the presence of intraoral hemangiomas. If they are present, patients should be referred to a pediatric dentist by age 1 year for early screening and management.
- Long-term outcomes and quality of life.
- An international group of experts published a report of a multicenter study that used cross-sectional interviews and chart review to examine long-term outcomes and quality of life for patients older than 10 years with PHACE syndrome.[68] Individuals were defined as having definite PHACE by previously reported guidelines.[47] This was the largest cohort of adolescents and adults with PHACE. Of 153 individuals who were contacted, 104 participated in the study (68%). The median age was 14 years (range, 10–77 years). This study found that PHACE syndrome was associated with long-term, mild-to-severe morbidities, including infantile hemangioma residua (94.1%), headaches/migraines (72.1%), learning differences (45.1%), and progressive arteriopathy (29.4%). Additional findings from the study are reported in Table 3.
Most patients with hemangioma residua were satisfied or very satisfied with their appearance (89.5%). Those with surgery and/or ulceration were less likely to report a minimal impact on self-confidence. Of the 68 patients with arteriopathy and available follow-up imaging, 6 (8.8%) developed moyamoya vasculopathy or progressive stenoocclusion, leading to isolated circulation at or above the level of the circle of Willis. Despite this finding, the proportion of patients with ischemic stroke was low (2 of 104; 1.9%). Patient-Reported Outcomes Measurement Information System (PROMIS) global health scores were lower than population norms by at least 1 standard deviation. Given the overall prevalence of PHACE, it was not possible to obtain the proper power to accurately assess all outcomes. The authors of the study concluded that primary and specialty follow-up care is important for patients with PHACE into adulthood. Further study is needed to identify precise guidelines for long-term follow-up.[68]
Table 3. Additional Findings Identified Among the PHACE Syndrome Cohorta Symptom Prevalence Symptom Prevalence ADHD = attention-deficit/hyperactivity syndrome; IH = infantile hemangioma. a Adapted from: Mitchell Braun, Ilona J. Frieden, Dawn H. Siegel, Elizabeth George, Christopher P. Hess, Christine K. Fox, Sarah L. Chamlin, Beth A. Drolet, Denise Metry, Elena Pope, Julie Powell, Kristen Holland, Caden Ulschmid, Marilyn G. Liang, Kelly K. Barry, Tina Ho, Chantal Cotter, Eulalia Baselga, David Bosquez, Surabhi Neerendranath Jain, Jordan K. Bui, Irene Lara-Corrales, Tracy Funk, Alison Small, Wenelia Baghoomian, Albert C. Yan, James R. Treat, Griffin Stockton Hogrogian, Charles Huang, Anita Haggstrom, Mary List, Catherine C. McCuaig, Victoria Barrio, Anthony J. Mancini, Leslie P. Lawley, Kerrie Grunnet-Satcher, Kimberly A. Horii, Brandon Newell, Amy Nopper, Maria C. Garzon, Margaret E. Scollan, Erin F. Mathes, Multicenter Study of Long-Term Outcomes and Quality of Life in PHACE Syndrome after Age 10, The Journal of Pediatrics, Volume 267, 2024, 113907, ISSN 0022-3476,https://doi.org/10.1016/j.jpeds.2024.113907. This is an open access article distributed under the terms of theCreative Commons CC-BY license, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. b Report of ever having had a seizure. c IncludingTourette syndrome,intention tremors, psychogenicmovement disorder. IH late growth 13/104 (12.5%) Vision difficulty 56/104 (53.8%) Increased color 11/13 (84.6%) Unilateral legal blindness 5/104 (4.8%) Deep growth 2/13 (15.4%) Eye surgeries 26/104 (25%) Increased volume 6/13 (46.2%) Hearing loss 18/104 (17.3%) Additional neurological symptoms Conductive 3/18 (16.7%) Seizuresb 15/104 (14.4%) Sensorineural 3/18 (16.7%) Speech difficulty 36/104 (34.6%) Mixed 6/18 (33.3%) Participated in speech therapy 30/104 (28.8%) Unknown 3/18 (16.7%) Balance problems 28/104 (26.9%) Use of hearing aids 12/104 (11.5%) Difficulty swallowing 11/104 (10.6%) Dental Tic disordersc 6/104 (5.8%) Dental root problem 16/104 (15.4%) Learning diagnosis Defects in enamel 31/104 (29.8%) ADHD 19/104 (18.3%) Dyslexia 10/104 (9.6%)
- An international group of experts published a report of a multicenter study that used cross-sectional interviews and chart review to examine long-term outcomes and quality of life for patients older than 10 years with PHACE syndrome.[68] Individuals were defined as having definite PHACE by previously reported guidelines.[47] This was the largest cohort of adolescents and adults with PHACE. Of 153 individuals who were contacted, 104 participated in the study (68%). The median age was 14 years (range, 10–77 years). This study found that PHACE syndrome was associated with long-term, mild-to-severe morbidities, including infantile hemangioma residua (94.1%), headaches/migraines (72.1%), learning differences (45.1%), and progressive arteriopathy (29.4%). Additional findings from the study are reported in Table 3.
LUMBAR/PELVIS/SACRAL syndrome
Infantile hemangiomas located over the lumbar or sacral spine may be associated with genitourinary, anorectal anomalies, or neurological issues such as tethered cord.[69,70,71,72] The following criteria have been used to describe segmental infantile hemangioma syndrome in the lumbar, pelvic, and sacral areas. This syndrome has been described in the literature using several acronyms.
LUMBAR
- L ower-body hemangiomas and other cutaneous defects.
- U rogenital anomalies or ulceration.
- M yelopathy.
- B ony deformities.
- A norectal malformations or arterial anomalies.
- R enal anomalies.
PELVIS
- P erineal hemangiomas.
- E xternal genital malformations.
- L ipomyelomeningocele.
- V esicorenal abnormalities.
- I mperforate anus.
- S kin tag.
SACRAL
- S pinal dysraphism.
- A nogenital.
- C utaneous.
- R enal and urologic anomalies A ssociated with an angioma of L umbosacral localization.
Segmental lesions over the gluteal cleft and lumbar spine need to be evaluated with either ultrasonography or MRI, depending on the age of the patient. In several studies, ultrasonography evaluations have failed to identify some spinal abnormalities that were later found on MRI evaluation.[73,74]
Multiple hemangiomas
Infants with more than five hemangiomas need to be evaluated for visceral hemangiomas. The most common site of involvement is the liver, in which multiple or diffuse lesions can be noted.[75,76,77] Often these lesions are asymptomatic, but in a minority of cases, symptoms such as heart failure secondary to large vessel shunts, compartment syndrome, or profound hypothyroidism can occur because of the expression of iodothyronine deiodinase by the hemangioma cells.[78] Multiple or diffuse liver hemangiomas can occur in the absence of skin lesions. Other rare potential complications of visceral hemangiomas depend on specific organ involvement and are caused by mass effects. These complications include gastrointestinal hemorrhage, obstructive jaundice, and CNS sequelae. For more information, see the Hepatic Vascular Tumors (HVT) section.
Treatment of infantile hemangioma
The decision to treat patients with hemangiomas is based on several factors, including the following:[79]
- Size of the lesions.
- Type of hemangioma.
- Location of hemangioma.
- Presence or risk of complications, including ulceration, possibility of scarring or disfigurement, the age of the patient, and the stage of growth of the hemangioma.
This decision is individualized among patients, and it is important to carefully consider the risks and benefits of treatment.
The American Academy of Pediatrics has published clinical practice guidelines on this topic. An early therapeutic intervention was noted to be critical for complex infantile hemangiomas to prevent medical complications and permanent disfigurement. The timing of interventions was noted to be best in the first 1 to 3 months of age. Photos were used to triage low-risk versus high-risk infantile hemangiomas,[80] and a scoring system was used for primary care physicians to encourage early referral to hemangioma specialists.[81] The guidelines indicated that hemangioma specialists are practitioners with expertise in the management and care of hemangiomas who have knowledge of risk stratification and treatment options. These providers consisted of experts in the fields of dermatology, hematology/oncology, pediatrics, plastic surgery, general surgery, otolaryngology, and ophthalmology.[82]
Treatment options for infantile hemangioma include the following:
- Propranolol therapy.
- Selective and other beta-blocker therapy.
- Corticosteroid therapy.
- Laser therapy.
- Usually reserved for ulcerated infantile hemangiomas and residual lesions, such as telangiectasias after the proliferative period.[83] Pulsed dye laser therapy helps with pain from ulcerative infantile hemangiomas. The use of pulsed dye laser therapy as an up-front treatment for infantile hemangiomas is controversial.
- A Russian pilot study employed multiline laser equipment using the Nd:YAP Q-Sw/KTP emitters combined with two wavelengths of 1079/540 nm to treat patients with infantile hemangiomas.[84] Laser treatment was performed on 109 patients with 119 hemangiomas. Evaluation of posttreatment samples revealed restoration of normal color, skin relief, and the absence of scars.
- A retrospective study in China included 180 patients with superficial hemangiomas who were treated with a 595-nm pulsed dye laser. The study reported that younger children (aged <2 months) received fewer treatments, had shorter courses of disease, and experienced better effects with fewer adverse events when compared with older children.[85]
- Excisional surgery. With the advent of new medical treatments, the use of surgery is reserved for ulcerated lesions, residual lesions, large periocular lesions that interfere with vision, and facial lesions with aesthetic impact that do not respond to medical therapy.[86]
- Topical beta-blocker therapy.
- Combined therapy for complicated hemangiomas.
Propranolol therapy
Propranolol, a nonselective beta-blocker, is first-line therapy for infantile hemangiomas. Early studies suggested that propranolol might act through inducing vasoconstriction and/or by decreasing expression of VEGF and bFGF, leading to apoptosis.[87,88] Subsequent studies indicate that the activity of propranolol for infantile hemangiomas is not secondary to beta blockade resulting from action of the S(-) enantiomer of propranolol but rather resulting from the ability of the R(+) enantiomer of propranolol to inhibit SOX18, a transcription factor that acts as a master regulator of vasculogenesis.[89,90,91] The R(+) enantiomer interferes with transcriptional activation by SOX18, disrupts SOX18-chromatin binding dynamics, and inhibits SOX18 dimer formation. These biochemical effects result in inhibition of hemangioma stem cell differentiation into endothelial cells and in inhibition of vasculogenesis.[91]
The use of propranolol was first noted in two infants treated for cardiac issues in Europe. A change in color, softening, and decrease in hemangioma size was noted. Since that time, the results of a randomized controlled trial have been reported.[92] In 2014, the U.S. Food and Drug Administration (FDA) approved Hemangeol, the pediatric formulation of propranolol hydrochloride, for the treatment of proliferating infantile hemangiomas. Generic propranolol remains in common use.
There are many other published reports about the efficacy and safety of propranolol.[93,94,95,96,97] Lack of response to treatment is rare. Propranolol therapy is usually used during the proliferative phase but has been effective in patients older than 12 months with infantile hemangiomas.[98]; [99][Level of evidence C3]
Evidence (propranolol therapy):
- In a large industry-sponsored randomized trial, 456 infants aged 5 weeks to 5 months with proliferating infantile hemangiomas of at least 1.5 cm received either a placebo or propranolol (1 mg/kg per day or 3 mg/kg per day) for 3 or 6 months. After interim analysis of the first 188 patients who completed 24 weeks of trial treatment, the regimen of 3 mg/kg per day for 6 months was selected for the final efficacy analysis.[92][Level of evidence B3]
- Of patients who received the selected regimen, 88% showed improvement by week 5, compared with 5% of patients who received the placebo.
- Adverse events occurred infrequently.
- A retrospective study of 635 infants with infantile hemangiomas who were treated with propranolol (2 mg/kg per day) had the following results:[97][Level of evidence C3]
- The overall response rate was 91%, with most patients demonstrating regression.
- Two percent of patients had side effects, none of which were severe.
- A meta-analysis that evaluated 5,130 patients from 61 studies concluded that propranolol was more effective and safer than were other treatments for infantile hemangioma.[100]
- Airway infantile hemangioma lesions are rare. A meta-analysis of 61 patients reported the following results:[101]
- There was a trend in decreased treatment failure with increased dosing strategies, which is consistent with the use of higher doses of propranolol in these patients (3 mg/kg per day).
- The analysis also suggested that the concurrent use of steroids and propranolol may have reduced efficacy in patients with segmental airway hemangiomas, but prior treatment with steroids had no deleterious effect.
- Additional prospective studies are needed to validate these findings.
- Diffuse (segmental) hemangiomas of the airway are very rare, and their clinical behavior is different from that of isolated airway lesions.
Intralesional administration of propranolol has been used for periorbital lesions in a limited capacity and showed no advantages over oral administration.[102][Level of evidence B3]
Several expert consensus panel recommendations have been reported, including recommendations from the FDA and the European Medicines Agency after a randomized controlled trial of oral propranolol in infantile hemangioma patients led to FDA approval.[103,104,105]
Considerations for the use of propranolol include the following:[103,105,106]
- Initiation of treatment: Guidance from consensus panels suggested that treatment should be undertaken in consultation with a pediatric vascular anomaly specialist with expertise in the diagnosis and treatment of pediatric vascular tumors and in the use of propranolol in children. They suggested that hospitalization for initiation of oral propranolol be considered in the following circumstances:[103]
- Infant aged 4 weeks or younger (corrected for gestational age).
- Infant of any age with inadequate social support.
- Infant of any age with comorbid conditions affecting the cardiovascular or respiratory system, including symptomatic airway infantile hemangiomas.
- Infant of any age with conditions affecting blood glucose maintenance.
The pretreatment evaluation (inpatient or outpatient) includes the following:
- History, with focus on cardiovascular and respiratory abnormalities (e.g., poor feeding, dyspnea, tachypnea, diaphoresis, wheezing, heart murmur) and family history of heart block or arrhythmia.
- Physical examination, including cardiac and pulmonary assessment and measurement of heart rate.
- No need for echocardiogram or electrocardiogram for standard-risk patients. Two studies found no contraindication to beta-blocker therapy in 6.5% to 25% of patients who had electrocardiogram abnormalities.[106,107] Electrocardiogram should be considered in children with heart rate lower than normal for age and history of arrhythmia or arrhythmia detected during examination.
- Family history of congenital heart disease or maternal history of connective tissue disease.
- Dosing: According to the consensus panels, the dosing used is generally 1 mg/kg per day to 3 mg/kg per day divided into two or three doses. The starting dose varies depending on risk factors and location of initiation. Outpatients and inpatients are initially started at a dose of 0.5 mg/kg per day to 1 mg/kg per day and increased over time.[104,105,106] A retrospective review of initial dosing indicates a starting dose of 2 mg/kg may also be well tolerated. This initial dosing could decrease the need for up-titration and more frequent clinic visits, although prospective studies are needed.[108] Initially, dosing of three times per day is recommended for infants younger than 5 weeks and for patients with PHACE syndrome.[47,103]
- Monitoring: Monitoring varies depending on the institution. However, oral propranolol peaks at 1 to 3 hours after administration and most centers measure heart rate and blood pressure 1 and 2 hours after each dose with initiation and then when the dose is increased by at least 0.5 mg/kg per day. Parent and patient education includes when to withhold the medication, signs of hypoglycemia, feeding necessity through the night, and when to call the physician with issues, such as illness, that may interfere with oral intake or lead to dehydration or respiratory problems.
A large retrospective multicenter study assessed the safety of outpatient administration of propranolol and evaluated the need for monitoring. In this study, 783 patients with 1,148 office visits were evaluated. No symptomatic bradycardia or hypotension was noted. Blood pressure evaluation was unreliable. The results suggested that outpatient evaluation may not be necessary for standard-risk patients with infantile hemangiomas.[109]
- Contraindications: Propranolol treatment is contraindicated in infants and children with the following:[103,104,105]
- Sinus bradycardia.
- Hypotension.
- Heart block greater than first degree.
- Heart failure.
- Asthma.
- Hypersensitivity.
- PHACE syndrome. PHACE syndrome with CNS arterial disease and/or coarctation of the aorta may be a relative contraindication. A retrospective multi-institutional study that investigated the safety of propranolol therapy for patients with PHACE syndrome identified 76 infants, including 12 patients who were at high risk of having a stroke.[110] The incidence of adverse events in these patients was similar to the incidence in 726 infants who received oral propranolol therapy for hemangioma but did not meet the criteria for PHACE syndrome. A decision to treat should be made in consultation with neurology/neurosurgery and cardiology.
- Adverse effects of propranolol include the following:[111]
- Hypoglycemia.
One study in Japan monitored hypoglycemia in infants with infantile hemangiomas who started treatment with propranolol.[112] After treatment with propranolol, the incidences of severe hypoglycemia and hypoglycemic convulsions were approximately 0.54% and 0.35%, respectively. The incidence of hypoglycemic convulsions appeared to be higher in Japan than in Western countries. Severe hypoglycemia was common in infants younger than 1 year when propranolol was used for 6 months or longer. Severe hypoglycemia often developed from 5:00 AM to 9:00 AM, and it was frequently associated with prolonged periods of fasting, poor feeding, or poor physical conditions.
- Hypotension.
- Bradycardia.
- Sleep disturbance.
- Diarrhea/constipation.
- Cold extremities.
These complications have been reported in several studies, and severe complications have been rare.[111,113] The risk of these complications is increased in patients with comorbidities and concomitant diseases, including diarrhea, vomiting, and respiratory infections. The need for close monitoring and possible periods of drug discontinuation should be considered during periods of illness.
A retrospective review of 1,260 children with infantile hemangiomas who were treated with propranolol identified 26 patients (2.1%) with side effects that required discontinuation of propranolol.[114] Severe sleep disturbance was the most common reason for propranolol cessation, accounting for 65.4% of cases. In total, 23 patients received atenolol and 3 patients received prednisolone as second-line therapy. In the multivariate analysis, only younger age (95% confidence interval [CI], 1.201–2.793; P = .009) and lower body weight (95% CI, 1.036–1.972; P = .014) were associated with intolerable side effects.
- Hypoglycemia.
- Duration of treatment: There are no consensus guidelines for the treatment duration of propranolol. In a prospective, multi-institutional study that assessed efficacy and safety of propranolol in high-risk patients, the administration of propranolol for a minimum of 6 months, up to a maximum of age 12 months, increased treatment success; dosing of propranolol was 3 mg/kg per day. Treatment results were sustained for up to 3 months after discontinuation of therapy. Efficacy and safety of propranolol in this study were similar to those reported in other studies.[115]
- Rebound growth after propranolol therapy: Rebound refers to the growth of infantile hemangiomas after propranolol cessation. A multi-institutional, retrospective review of 997 patients with infantile hemangiomas found a rebound rate of 25.3% in 912 patients with adequate data. On univariate analysis, the factors associated with rebound included discontinuation of treatment before age 9 months, female sex, location on the head or neck, segmental pattern, and deep or mixed skin involvement. On multivariate analysis, only deep infantile hemangiomas and female sex were significantly related.[116] A single-center retrospective review examined 198 patients with infantile hemangioma who underwent oral propranolol therapy. The study reported 35 patients (18%) with rebound growth 1 to 3 months after discontinuation of propranolol treatment. Of the 35 patients, 23 were re-treated with propranolol for up to 3 months. All patients had good responses.[117][Level of evidence C3]
- Late growth of infantile hemangiomas: Hemangioma growth can occur in patients older than 3 years, and growth as late as age 8.5 years has been reported. Associated risk factors include segmental morphology, large hemangiomas, PHACE syndrome, and deep cutaneous and subcutaneous lesions in the head and neck.[118,119]
Selective and other beta-blocker therapy
Because of the nonselective and lipophilic nature of propranolol and its ability to cross the blood-brain barrier, other beta-blockers are being used for the treatment of infantile hemangiomas.
Evidence (beta-blocker therapy):
- In two small comparison studies, there was no difference in efficacy between propranolol and atenolol.[120,121]
- In support of a previous retrospective study, a prospective double-blind study compared nadolol with propranolol in 71 infants (aged 1–6 months).[122][Level of evidence A3]
- The study demonstrated noninferiority with respect to efficacy and treatment.
- A prospective study of 76 infants treated with atenolol noted efficacy and safety similar to propranolol.[123][Level of evidence C3]
In one published report, nadolol was associated with the death of an infant (aged 17 weeks) after 10 days of no stool output.[124] There is limited information about the pharmacokinetics and safety of nadolol in infants. The drug has a narrow therapeutic index, and it is excreted and remains unchanged in the feces. If an infant is given nadolol, it is critical to monitor for regular stool output.
Additional studies are needed to assess differences between the toxicities of these agents and the toxicities of propranolol.
There is some suggestion that the more selective beta-blockers have fewer side effects.[125] A study has suggested that the R(+) enantiomer of propranolol, carried over in drug synthesis rather than the anti–beta-adrenergic L(-) enantiomer (commercially available drug is a racemic mixture), may carry the therapeutic anti-infantile hemangioma effect.[89,90]
Corticosteroid therapy
Before propranolol, corticosteroids were the first line of treatment for infantile hemangiomas. They were first used in the late 1950s but were never approved by the U.S. FDA for this indication. Corticosteroid therapy has become less popular because of the acute and long-term side effects of steroids (gastrointestinal irritability, immunosuppression, adrenocortical suppression, cushingoid features, and growth failure).
Corticosteroids (prednisone or methylprednisolone) are used at times when there is a contraindication to beta-blocker therapy or as initial treatment while a patient is started on beta-blocker therapy.[126]
Topical beta-blocker therapy
Topical beta-blockers are used mainly for the treatment of small, localized, superficial hemangiomas as an alternative to observation. They have also been used in combination with systemic therapy in complicated hemangiomas or to prevent rebound in hemangiomas being tapered off of systemic treatment.[127,128,129] The same precautions (assessment of comorbidities and family history), as noted previously for propranolol, should be followed for topical beta-blockers. Systemic absorption (plasma and urine) of timolol is variable and prescreening for normal cardiac, pulmonary, and endocrine issues are essential. Recent medical histories and physical examinations are also important. Cautious administration is necessary for ulcerated and deep hemangiomas because higher plasma concentrations of timolol can be seen.[130,131]
The topical timolol that is used is the ophthalmic gel-forming solution 0.5%. One drop is applied to the hemangioma two times per day until stable response is achieved.
This treatment has limited side effects, but infants with a postmenstrual age of younger than 44 weeks and weight at treatment initiation of less than 2,500 grams may be at risk of adverse events, including bradycardia, hypotension, apnea, and hypothermia.[131,132] Close monitoring of temperature, blood pressure, and heart rate in premature and low birth weight infants with infantile hemangiomas at initiation of and during therapy with topical timolol is necessary.
Evidence (topical timolol therapy):
- A retrospective cohort study included 666 patients with infantile hemangioma who were treated with topical timolol for 12 months.[133]
- Of these patients, 583 (87.5%) had visible reductions in the size of their lesions.
- A total of 188 children (28.2%) had excellent responses (no remaining visible abnormality), 127 of whom had complete responses earlier than 12 months.
- Of the remaining patients, 292 (43.8%) had good outcomes (i.e., the hemangioma was less than half its original size), while 103 (15.5%) had fair outcomes (i.e., a visibly smaller hemangioma but not less than half its original size), and 83 (12.5%) had poor outcomes (i.e., there was no change in hemangioma size or it was larger).
- Patients aged 3 months and younger were more likely to have better outcomes than those older than 3 months (P < .001).
- Patients with small infantile hemangiomas (maximum diameter, 1.5–≤5 cm) also had better outcomes than those with large infantile hemangiomas (5–≤10 cm) (P = .046).
- In a multicenter, retrospective, cohort study, 731 children with predominantly superficial hemangiomas were treated with topical timolol 0.5% twice daily.[129]
- Ninety-two percent of patients showed significant improvement in hemangioma color.
- Seventy-seven percent of patients showed improvement in hemangioma size, extent, and volume.
- Topical timolol is generally well tolerated; however, data on its safety are limited.
- A Spanish consortium performed a prospective randomized trial to evaluate the efficacy and safety of topical timolol for the treatment of infantile hemangioma in the early proliferative stage.[134] This multicenter, randomized, double-blind, placebo-controlled, phase IIa pilot clinical trial included patients aged 10 to 60 days with focal or segmental hemangiomas (superficial, deep, mixed, or minimal/arrested growth). Patients were randomly assigned to treatment with either topical timolol maleate solution, 0.5%, or placebo, twice daily for 24 weeks.
- At 24 weeks, there were no significant differences between the timolol treatment and the placebo for complete or nearly complete infantile hemangioma resolution (42% for timolol [n = 11] vs. 36% for placebo [n = 11]; P = .37).
Combined therapy for complicated hemangiomas
Combined therapy is considered either at initiation of treatment in complicated lesions in which there is functional impairment or organ compromise or used at the end of systemic therapy to prevent hemangioma rebound. Further investigation of efficacy and safety is needed for these regimens.
Evidence (combined therapy for complicated hemangiomas):
- A prospective randomized study that compared propranolol and 2 weeks of steroid therapy with propranolol alone revealed the following:[135]
- Decreased sizes of hemangiomas at 2, 4, and 8 weeks in the combined-therapy group but no statistical difference in the sizes at 6 months.
- A prospective randomized study that compared timolol and propranolol with propranolol alone reported the following:[136]
Treatment options under clinical evaluation for infantile hemangiomas
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
In response to the COVID-19 pandemic, the Hemangioma Investigator Group is studying the administration of propranolol for low-risk and standard-risk patients through virtual visits.[139]
Current Clinical Trials
Use our advanced clinical trial search to find NCI-supported cancer clinical trials that are now enrolling patients. The search can be narrowed by location of the trial, type of treatment, name of the drug, and other criteria. General information about clinical trials is also available.
Congenital Hemangiomas
Clinical features and diagnostic evaluation
Congenital hemangiomas can be difficult to diagnose, especially for clinicians who are unfamiliar with these lesions. Diagnostic criteria include a purpuric lesion fully formed at birth, frequently with a halo around the lesion, with high flow noted on ultrasound imaging. Essential to the diagnosis is serial observation for decrease or, at least stability, in size over time. These lesions do not enlarge unless there is hemorrhage into the tumor.
Congenital hemangiomas are divided into the following three forms:
- Rapidly involuting congenital hemangiomas (RICH). These lesions are large high-flow lesions that are completely formed at birth but rapidly involute by age 12 to 15 months. They can ulcerate and bleed and can cause transient heart failure and mild coagulopathy. After involution, usually some residual changes in the skin are present (see Figure 5).[140,141,142,143]
In a retrospective case series of congenital hemangiomas, several high-risk ultrasound findings were noted for RICH. Venous lakes were associated with cardiac failure, and an increased risk of bleeding was noted with venous lakes and venous ectasia. Infants with RICH should be evaluated with ultrasonography and monitored closely if these high-risk features are noted.[144]
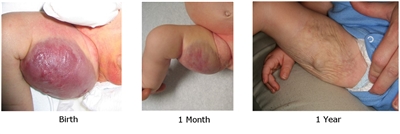
Figure 5. Typical appearance of a cutaneous congenital hemangioma at birth. Note the pedunculated mass. This RICH lesion involuted over time but some residual skin changes remained. Credit: Denise Adams, M.D. - Partial involuting congenital hemangiomas (PICH). These lesions are completely formed at birth and involute only partially.[145]
- Non-involuting congenital hemangiomas (NICH). These lesions are formed at birth and never involute. Depending on the location of the lesions and whether they cause functional impairment, the lesions may need to be removed surgically.[146,147]
Histopathology and molecular features
Congenital hemangiomas are benign vascular tumors that proliferate in utero. Development of these lesions is complete at birth. Histologically, these lesions are GLUT1 negative, unlike infantile hemangiomas. They are usually cutaneous, but can be found in the viscera. Complications include hemorrhage, transient heart failure, and transient coagulopathy.[148]
Somatic activating variants of GNAQ and GNA11 have been found to be associated with congenital hemangiomas.[149] Additional research is necessary to assess the significance of these findings, as this may aid in diagnosis and pathophysiology.
Hepatic Vascular Tumors
With the development of the new WHO and ISSVA classifications, the terminology of pediatric hepatic vascular tumors (HVT) has changed.[150,151,152] The historical Dehner classification of types 1, 2, and 3 liver hemangioendothelioma is no longer favored by pathologists.[153] The term hemangioendothelioma is not considered an isolated entity.
On MRI, vascular tumors of the liver are hyperintense on T2 imaging and hypointense on T1 imaging, with postcontrast imaging demonstrating early peripheral enhancement with eventual diffuse enhancement.[76] In practice, these tumors have been classified according to their clinical characteristics and radiological assessment.[76,154] In general, hepatic vascular tumors can be benign or malignant.
The differential diagnosis of vascular liver lesions always includes malignant liver tumors. Thus, alpha-fetoprotein (AFP) measurements should be included in the initial lab work. AFP is very high in all newborns but will rapidly fall to normal levels in several months. AFP levels should rapidly diminish, but failure to do so or a rising trend of AFP should elicit concern for a hepatoblastoma. There are no prospective studies investigating AFP elevation in patients with hemangiomas.[155,156] Some hypervascular hepatoblastomas in neonates with congestive heart failure have been mistaken for infantile hemangiomas. Other tumors in the differential diagnosis include angiosarcomas, metastatic neuroblastomas, and mesenchymal hamartomas. If there is any question about the diagnosis, a biopsy is recommended, although bleeding is a risk of the procedure.[157]
Benign hepatic vascular tumors
These lesions are usually divided into the following three categories:[76,154]
A more appropriate classification uses an interdisciplinary evaluation, including pathological classification with genomic assessment, radiological imaging evaluation, and clinical history and examination. This is based on the ISSVA and WHO classifications. A study analyzed clinicopathologic characteristics in 33 cases of pediatric hepatic vascular tumors diagnosed between 1970 and 2021.[158] Thirteen cases were identified as hepatic congenital hemangiomas. All were single lesions, and most of these were RICH. Ten patients had hepatic infantile hemangiomas. Three patients had hepatic angiosarcoma, and one patient had hepatic epithelioid hemangioendothelioma. Six patients were excluded from the study, five with vascular malformations and one with vascular dominant mesenchymal hamartoma. The study revealed the importance of an interdisciplinary team approach in the assessment of these tumors.
Congenital hemangiomas
Focal lesions of the liver are usually congenital hemangiomas (RICH or NICH, rarely PICH) (see Figure 6). RICH can present with symptoms of heart failure and mild to moderate coagulopathy but are typically detected by antenatal ultrasonography or as an asymptomatic mass in the newborn period.
Treatment options for focal vascular lesions of the liver include the following:
- Supportive management. Most lesions are asymptomatic and can be monitored through involution using ultrasonography.
- Embolization. This procedure is considered for severe symptomatic shunting that is unresponsive to treatment for congestive heart failure. These procedures need to be performed by interventional radiologists with expertise in vascular anomalies.[159]
- Surgery. Patients with massive focal symptomatic hepatic congenital hemangioma unresponsive to supportive management or radiological intervention may be candidates for surgical resection. This is a rare circumstance and needs to be evaluated by an interdisciplinary vascular anomaly team. Indication for surgical removal includes rupture, bleeding, and nonresolving coagulopathy. Two patients were reported to require surgical resection after the development of clinically significant ascites as their RICH involuted.[160,161]
No medication has proven to be an effective treatment for these lesions, and infants need to be supported during the initial period until involution begins.[76,154] These lesions may be diagnosed prenatally. In rare situations, maternal treatment with medications such as steroids appeared to be effective but, more likely, natural involution may have been responsible.[162]
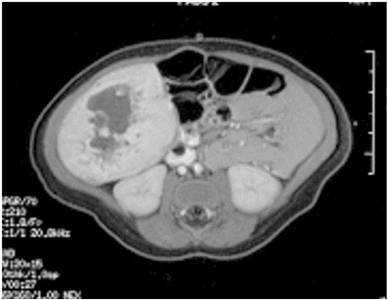
Figure 6. Single liver lesion (intrahepatic congenital hemangioma). MRI image of a congenital hemangioma. Note the central enhancement, which is typical for an intrahepatic congenital hemangioma. Credit: Denise Adams, M.D.
Infantile hemangiomas
Multifocal hepatic lesions are infantile hemangiomas. Multifocal lesions may not need to be treated if the patient is asymptomatic. These lesions typically follow the same proliferative and involution course as cutaneous hemangiomas.[76,154] These lesions are monitored closely and if there is growth, propranolol therapy should be considered. If propranolol is needed, doses of up to 2 mg/kg per day are effective.
Diffuse hepatic infantile hemangiomas
Diffuse liver lesions are very serious (see Figure 7). Complications include hypothyroidism caused by the expression of iodothyronine deiodinase, high-output or congestive heart failure, and abdominal compartment syndrome.[75,76,163,164]
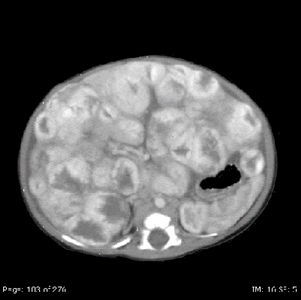
Figure 7. Diffuse liver lesions with classical imaging on CT. Note the peripheral enhancement in early contrast phase. Credit: Denise Adams, M.D.
Treatment options for diffuse liver lesions may include the following:
- Propranolol: Beta-blockers are the most common treatment for diffuse and some multifocal infantile hemangiomas of the liver. Treatment doses of 2 to 3 mg/kg per day are indicated.[92]
- Thyroid hormone replacement: Thyroid hormone replacement therapy must be aggressive if hypothyroidism is diagnosed. Treatment with higher doses of hormones may be needed because the deficiency is caused by the aggressive consumption of the hormone by the tumor.[78]
- Chemotherapy: Steroids, cyclophosphamide, and vincristine have been used to treat diffuse liver infantile hemangioma.[76,165,166]
- Liver transplant: If a patient does not respond to medical management, a transplant may be indicated.[167] Transplant is considered only for patients with severe diffuse lesions who have multisystem organ failure and there is insufficient time for effective pharmacologic therapy.
Malignant hepatic vascular tumors
There have been isolated reports of malignancy in patients with diffuse hepatic infantile hemangiomas.[168,169] It is not clear that all cases were caused by the transformation of a benign lesion to a malignant phenotype. However, if the lesion does not respond to standard therapy, biopsy should be considered. Further evaluation and consensus is needed to assess whether these patients need to be monitored over a longer period of time with liver ultrasonography. For more information, see the Angiosarcoma section.
Hepatic angiosarcoma
Hepatic angiosarcoma (HA) in children is extremely rare, and there are approximately 80 cases reported in the medical literature. There is a female predilection, and the median age at diagnosis is 40 months. Hepatic angiosarcomas present rapidly and are diagnosed by histopathology (see Table 4). These malignant tumors are treated with chemotherapy, embolization, antiangiogenic agents, and liver transplant (for those without metastases). Patients have a poor prognosis.[170] For more information, see the Angiosarcoma section.
Hepatic epithelioid hemangioendothelioma
Hepatic epithelioid hemangioendothelioma is a very rare tumor in the liver, especially in children. There is also a female predilection, and it occurs most frequently in young adults. This tumor may often involve extrahepatic disease, reportedly in over one-third of patients. It presents similarly to other hepatic tumors, with hepatomegaly and abdominal pain. However, hepatic epithelioid hemangioendothelioma behaves more moderately than hepatic angiosarcoma, and patients with hepatic epithelioid hemangioendothelioma have better outcomes.
Imaging is very helpful in diagnosing hepatic epithelioid hemangioendothelioma. In addition to a hypoechoic lesion on ultrasonography, ultrasonography with contrast or MRI with contrast show a typical target sign, due to concentric filling of the tumor. This is thought to be due to concentric areas of necrosis and alternating areas of dense, active tumor cells. In addition, hepatic epithelioid hemangioendothelioma is known for avid glucose uptake. The use of fluorine F 18-fludeoxyglucose (18F-FDG) positron emission tomography (PET), with computed tomography (CT) or MRI (PET-CT/PET-MRI), is helpful in confirming the diagnosis and determining organ involvement since these tumors often have extrahepatic disease. This imaging modality is also helpful in monitoring patients for disease recurrence after intervention. Ultimately, histopathological diagnosis is the preferred method, and these tumors exhibit both epithelioid and histiocytic/dendritic cells and a characteristic immunohistochemistry pattern in tumor samples (see Table 4).[171]
Treatment of hepatic epithelioid hemangioendothelioma is primarily surgical resection, either hepatectomy or liver transplant. Interventional procedures can also be used. Studies are evaluating whether incorporating chemotherapy into the treatment plan improves outcomes.[171]
| Features | Hepatic Congenital Hemangioma (HCH) | Hepatic Infantile Hemangioma (HIH) | Hepatic Angiosarcoma (HA) | Hepatic Epithelioid Hemangioendothelioma (HEHE) |
|---|---|---|---|---|
| CHF = congestive heart failure; NICH = noninvoluting congenital hemangioma; PICH = partially involuting congenital hemangioma; RICH = rapidly involuting congenital hemangioma. | ||||
| a Adapted from Berklite et al.[158] | ||||
| Clinical Presentation | Noted at birth or prenatally; CHF; transient coagulopathy; single lesions; RICH, PICH, rarely NICH | Noted postnatally, usually associated with skin lesions; diffuse lesions with significant hypothyroidism and CHF | Rare in pediatrics, has been seen in neonates and toddlers; very aggressive | Very rare; associated with other lesions (bone, lung); variable course |
| Imaging | Solid lesion | Multiple or diffuse lesions | Large infiltrative, can be multiple or diffuse, can be seen with IH, or rarely transformation can occur to HA | Solid or multiple lesions |
| Histology | Involutional changes (calcification, necrosis), dilated, fibrotic stroma capillary vessels | Anastomosing sinusoidal vasculature, dense normal appearing endothelial cells | Marked cytological atypia, infiltrative, epithelioid to spindle tumor cells, marked mitotic activity | Epithelioid endothelial cells in a background of myxohyaline stroma |
| GLUT1 | Negative | Positive | Positive in 20% of tumors | Negative |
| Somatic Variants or Gene Fusions | GNAQ,GNA11 | None | KRAS,KDR,PTPRB,FLT4,PLCG1,PIK3CA,TP53,TIE1,AKT1,CIC | YAP1::TFE3,WWTR1::CAMTA1 |
Spindle Cell Hemangioma
Clinical presentation, molecular features, and histopathology
Spindle cell hemangiomas, initially called spindle cell hemangioendotheliomas, often occur as superficial (skin and subcutis), painful lesions involving distal extremities in children and adults.[172,173] The tumors appear as red-brown or bluish lesions that can begin as a single nodule and develop into multifocal painful lesions over years. The hemangiomas are well circumscribed, occasionally contain phleboliths, and consist of cavernous blood spaces alternating with areas of nodular spindle cell proliferation. A significant percentage of spindle cell hemangiomas are completely intravascular. The vein containing the tumor is abnormal, as are blood vessels apart from the tumor mass.[174,175]
Spindle cell hemangiomas can be seen in patients with Maffucci syndrome (cutaneous spindle cell hemangiomas occurring with cartilaginous tumors, enchondromas) and Klippel-Trénaunay syndrome (capillary/lymphatic/venous malformations), generalized lymphatic anomalies, lymphedema, and organized thrombus.[174,175] In Maffucci syndrome, spindle cell hemangiomas are associated with IDH1 or IDH2 variants.[176]
Treatment of spindle cell hemangioma
There is no standard treatment for spindle cell hemangioma because it has not been studied in clinical trials. Surgical removal is usually curative, although there is a risk of recurrence.[174,175]
Epithelioid Hemangioma
Clinical presentation and histopathology
Epithelioid hemangiomas (EH) are benign lesions that usually occur in the skin and subcutis but can occur in other areas such as the bone, with focal and multifocal lesions.[174,177] Epithelioid hemangiomas may be a reactive process, as they can be associated with local trauma and can develop in pregnancy. Patients usually present with local swelling and pain at the involved site. In the bone, they present as well-defined lytic lesions that involve the metaphysis and diaphysis of long bones.[174,178] They can have a mixed lytic and sclerotic pattern of bone destruction.
On pathological evaluation, epithelioid hemangiomas have small caliber capillaries with eosinophilic, vacuolated cytoplasm and large oval, grooved, and lobulated nuclei. The endothelial cells are plump and are mature, well-formed vessels surrounded by multiple epithelioid endothelial cells within abundant cytoplasm. They lack cellular atypia and mitotic activity.[174,177,178,179]
In a study of 58 cases of epithelioid hemangiomas, 29% were found to have FOS gene rearrangements. FOS gene rearrangements were noted more often in cellular epithelioid hemangiomas and intraosseous lesions compared with lesions in the skin, soft tissue, and head and neck. This genetic abnormality can be helpful in distinguishing epithelioid hemangiomas from other malignant epithelioid vascular tumors.[179]
A single-institution report reviewed 11 patients with epithelioid hemangiomas (median age, 14.4 years) who were diagnosed between 1999 and 2017. Lesions occurred in the lower extremities (five patients), skull (three patients), pelvis (two patients), and spine (one patient). Five patients had multifocal disease. Patients presented with localized pain and neurological symptoms, including cranial nerve injury. No significant cytological atypia was noted, and the endothelial cells were positive for CD31 and ERG, and negative for cytokeratin and CAMPTA1. Median follow-up was 1.5 years. Various modalities of treatments were used, including surgery, endovascular embolization, cryoablation, and medical management. One patient received sirolimus, and another patient received interferon; the lesions of both patients shrank within the first year of follow-up. The youngest patient, aged 2.5 years, had multifocal skull lesions that partially regressed 1 year later without treatment.[180]
Treatment of epithelioid hemangioma
There is no standard treatment for epithelioid hemangioma because it has not been studied in clinical trials. Treatment usually consists of curettage, sclerotherapy, or resection. In rare cases, radiation therapy may be used.[174,178]
Pyogenic Granuloma (Lobular Capillary Hemangioma)
Clinical presentation, histopathology, and molecular features
Pyogenic granulomas (PG), known as lobular capillary hemangiomas, are benign reactive lesions. Pyogenic granulomas can present at any age—including at birth (congenitally), during the neonatal period, during infancy, or during pregnancy—although they are most common in older children and young adults. These lesions can arise spontaneously, in sites of trauma, or within capillary and arteriovenous malformations. Pyogenic granulomas have also been associated with medications including oral contraceptives and retinoids.
Pyogenic granulomas occur as solitary growths, but multiple (grouped) or rarely disseminated lesions have been described.[181] These lesions appear as small or large, smooth or lobulated vascular nodules that can grow rapidly, sometimes over weeks to months and have a tendency to bleed profusely. These lesions are usually cutaneous, but deep-seated/subcutaneous pyogenic granulomas have been reported and mimic other vascular lesions.[182] Histologically, these lesions are composed of capillaries and venules with plump endothelial cells separated into lobules by fibromyxoid stroma. Some untreated lesions eventually atrophy, become fibromatous, and slowly regress. A retrospective review of a series of eight children with disseminated congenital or neonatal pyogenic granulomas reported the occurrence of hemorrhagic central nervous system lesions in seven patients, five of whom developed neurological sequelae. Four of the eight patients had transient coagulopathy.[183][Level of evidence C2]
The pathogenesis of pyogenic granulomas associated with capillary malformations and those that are sporadic are unknown. A study investigated ten patients with pyogenic granulomas arising from a capillary malformation and found eight with BRAF c.1799T>A variants, one with an NRAS c.182A>G variant, and one with a GNAQ c.548G>A variant. This GNAQ variant was also found in the underlying capillary malformation. In 25 patients with pyogenic granulomas and no capillary malformation, 3 patients had BRAF c.1799T>A variants and 1 patient had a KRAS c.37G>C variant. These genetic findings will help with future treatment modalities for this benign vascular tumor.[184]
Treatment of pyogenic granuloma
Full-thickness excision is the treatment with the lowest recurrence rate (around 3%),[185] but curettage, laser photocoagulation, or cryotherapy can also be used.[186] Topical timolol and propranolol have also been used.
Evidence (topical beta-blockers):
- In a single-arm series of patients with acquired ocular pyogenic granulomas, a small number of pediatric patients were treated for 21 days to 6 weeks with twice-daily topical timolol, 0.5%.[187,188][Level of evidence C3]
- Complete or near-complete responses without subsequent recurrence or progression were noted in 75% to 100% of the patients (all ages).
- A study of 22 patients with cutaneous pyogenic granulomas who were treated with topical 1% propranolol ointment with occlusion had the following results:[189]
- Fifty-nine percent of patients achieved complete responses (mean, 66 days), 18% of patients had stable disease, and 22% of patients did not respond to the treatment.
- In this study, only skin toxicity was assessed.
- The authors did not comment on the penetrance of the propranolol formulation or include a safety evaluation of the side effects such as hypoglycemia and the effects on heart rate or blood pressure.
Angiofibroma
Clinical presentation
Angiofibromas are rare, benign neoplasms in the pediatric population. Typically, they are cutaneous lesions associated with tuberous sclerosis, appearing as red papules on the face.
Treatment of angiofibroma
Excision of the tumor, laser treatments, and topical treatments, such as sirolimus, have been used.[190,191,192]
Evidence (topical sirolimus):
- A prospective, randomized, placebo-controlled trial of nine cancer centers that included 62 patients who received sirolimus gel demonstrated the following results:[193]
- Sixty percent of patients who received sirolimus showed significant improvement in the size and color of the lesion, which was assessed at week 12.
- In another prospective, multicenter, randomized, double-blind, vehicle-controlled study that included six monthly clinic visits, 179 patients with tuberous sclerosis complex–related facial angiofibromas were treated with topical rapamycin (sirolimus) 0.3 g per 30 g (1%).[194]
- According to the Angiofibroma Grading Scale, patients who were treated with topical rapamycin showed a statistically and clinically significant improvement in facial angiofibromas.
Juvenile Nasopharyngeal Angiofibroma
Clinical presentation and histopathology
Juvenile nasopharyngeal angiofibromas (JNA) account for 0.5% of all head and neck tumors.[195] They typically occur in peri-pubertal males. While juvenile nasopharyngeal angiofibromas have not classically been included among vascular tumors, histologically, these tumors appear to be vascular tumors, with cells expressing vascular endothelial marker CD31, VEGFA, and VEGFR1.
Despite their benign-appearing histology, juvenile nasopharyngeal angiofibromas can be locally destructive, spreading from the nasal cavity to the nasopharynx, paranasal sinuses, and orbit skull base, with intracranial extension. Some publications have suggested a hormonal influence on juvenile nasopharyngeal angiofibromas, with emphasis on the molecular mechanisms involved.[196,197] Nineteen patients with clinico-radiologically diagnosed primary juvenile nasopharyngeal angiofibromas underwent gallium Ga 68-[DOTA, 1-Nal3]-octreotide (68Ga-DOTANOC) PET-CT scans.[198] The rationale for using this scan was the high expression of somatostatin receptors (SSTRs) in these tumors. DOTANOC expression was noted in all 19 cases of primary juvenile nasopharyngeal tumors (100%). The mean DOTANOC maximum standardized uptake value ratio of tumor and background was 6.9 (±1.4) (range, 3.8–9.5). Intracranial extension in 13 of 19 patients was prominently visualized because of the absence of DOTANOC uptake in the brain. The authors suggested that these findings open possibilities for physiological diagnostic imaging, with a promise of greater specificity and sensitivity. This scan may be applicable in ambivalent diagnostic situations, such as the detection of recurrence.
Treatment of juvenile nasopharyngeal angiofibroma
Surgical excision is the treatment of choice, but this can be challenging because of the extent of the lesion. A single-institution retrospective review of juvenile nasopharyngeal angiofibromas identified 37 patients with lateral extension.[199] Anterior lateral extension to the pterygopalatine fossa occurred in 36 patients (97%) and further to the infratemporal fossa in 20 patients (54%). In 16 patients (43%), posterior lateral spread was observed (posterior to the pterygoid process and/or between its plates). The recurrence rate was 29.7% (11 of 37 patients). The recurrence rate in patients with anterior and/or posterior lateral extension was significantly higher than in patients with anterior lateral extension only.
Juvenile nasopharyngeal angiofibromas have also been treated with radiation therapy, chemotherapy, alpha-interferon therapy, and sirolimus.[200,201,202,203,204]
References:
- Kilcline C, Frieden IJ: Infantile hemangiomas: how common are they? A systematic review of the medical literature. Pediatr Dermatol 25 (2): 168-73, 2008 Mar-Apr.
- Munden A, Butschek R, Tom WL, et al.: Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol 170 (4): 907-13, 2014.
- Darrow DH, Greene AK, Mancini AJ, et al.: Diagnosis and Management of Infantile Hemangioma. Pediatrics 136 (4): e1060-104, 2015.
- Darrow DH, Greene AK, Mancini AJ, et al.: Diagnosis and Management of Infantile Hemangioma: Executive Summary. Pediatrics 136 (4): 786-91, 2015.
- Haggstrom AN, Drolet BA, Baselga E, et al.: Prospective study of infantile hemangiomas: demographic, prenatal, and perinatal characteristics. J Pediatr 150 (3): 291-4, 2007.
- Dickison P, Christou E, Wargon O: A prospective study of infantile hemangiomas with a focus on incidence and risk factors. Pediatr Dermatol 28 (6): 663-669, 2011.
- Chen XD, Ma G, Chen H, et al.: Maternal and perinatal risk factors for infantile hemangioma: a case-control study. Pediatr Dermatol 30 (4): 457-61, 2013.
- Chang LC, Haggstrom AN, Drolet BA, et al.: Growth characteristics of infantile hemangiomas: implications for management. Pediatrics 122 (2): 360-7, 2008.
- Tollefson MM, Frieden IJ: Early growth of infantile hemangiomas: what parents' photographs tell us. Pediatrics 130 (2): e314-20, 2012.
- Haggstrom AN, Lammer EJ, Schneider RA, et al.: Patterns of infantile hemangiomas: new clues to hemangioma pathogenesis and embryonic facial development. Pediatrics 117 (3): 698-703, 2006.
- Waner M, North PE, Scherer KA, et al.: The nonrandom distribution of facial hemangiomas. Arch Dermatol 139 (7): 869-75, 2003.
- Chiller KG, Passaro D, Frieden IJ: Hemangiomas of infancy: clinical characteristics, morphologic subtypes, and their relationship to race, ethnicity, and sex. Arch Dermatol 138 (12): 1567-76, 2002.
- Metry DW, Garzon MC, Drolet BA, et al.: PHACE syndrome: current knowledge, future directions. Pediatr Dermatol 26 (4): 381-98, 2009 Jul-Aug.
- Munabi NC, Tan QK, Garzon MC, et al.: Growth Hormone Induces Recurrence of Infantile Hemangiomas After Apparent Involution: Evidence of Growth Hormone Receptors in Infantile Hemangioma. Pediatr Dermatol 32 (4): 539-43, 2015 Jul-Aug.
- Olsen GM, Nackers A, Drolet BA: Infantile and congenital hemangiomas. Semin Pediatr Surg 29 (5): 150969, 2020.
- Haggstrom AN, Drolet BA, Baselga E, et al.: Prospective study of infantile hemangiomas: clinical characteristics predicting complications and treatment. Pediatrics 118 (3): 882-7, 2006.
- Braun M, Metry D, Frieden IJ, et al.: Persistent dysesthesias in involuted infantile hemangiomas: An uncommon complication in a common condition. Pediatr Dermatol 38 (5): 1061-1065, 2021.
- Baselga E, Roe E, Coulie J, et al.: Risk Factors for Degree and Type of Sequelae After Involution of Untreated Hemangiomas of Infancy. JAMA Dermatol 152 (11): 1239-1243, 2016.
- Blei F, Walter J, Orlow SJ, et al.: Familial segregation of hemangiomas and vascular malformations as an autosomal dominant trait. Arch Dermatol 134 (6): 718-22, 1998.
- Castrén E, Salminen P, Vikkula M, et al.: Inheritance Patterns of Infantile Hemangioma. Pediatrics 138 (5): , 2016.
- Greenberger S, Bischoff J: Pathogenesis of infantile haemangioma. Br J Dermatol 169 (1): 12-9, 2013.
- Biswas A, Richards JE, Massaro J, et al.: Mast cells in cutaneous tumors: innocent bystander or maestro conductor? Int J Dermatol 53 (7): 806-11, 2014.
- Khan ZA, Boscolo E, Picard A, et al.: Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest 118 (7): 2592-9, 2008.
- Boye E, Yu Y, Paranya G, et al.: Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107 (6): 745-52, 2001.
- Yu Y, Flint AF, Mulliken JB, et al.: Endothelial progenitor cells in infantile hemangioma. Blood 103 (4): 1373-5, 2004.
- Boscolo E, Mulliken JB, Bischoff J: Pericytes from infantile hemangioma display proangiogenic properties and dysregulated angiopoietin-1. Arterioscler Thromb Vasc Biol 33 (3): 501-9, 2013.
- Zhang H, Wei T, Johnson A, et al.: NOTCH pathway activation in infantile hemangiomas. J Vasc Surg Venous Lymphat Disord 9 (2): 489-496, 2021.
- Barnés CM, Huang S, Kaipainen A, et al.: Evidence by molecular profiling for a placental origin of infantile hemangioma. Proc Natl Acad Sci U S A 102 (52): 19097-102, 2005.
- Walter JW, North PE, Waner M, et al.: Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer 33 (3): 295-303, 2002.
- Ritter MR, Dorrell MI, Edmonds J, et al.: Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Natl Acad Sci U S A 99 (11): 7455-60, 2002.
- Takahashi K, Mulliken JB, Kozakewich HP, et al.: Cellular markers that distinguish the phases of hemangioma during infancy and childhood. J Clin Invest 93 (6): 2357-64, 1994.
- Ritter MR, Reinisch J, Friedlander SF, et al.: Myeloid cells in infantile hemangioma. Am J Pathol 168 (2): 621-8, 2006.
- Bielenberg DR, Bucana CD, Sanchez R, et al.: Progressive growth of infantile cutaneous hemangiomas is directly correlated with hyperplasia and angiogenesis of adjacent epidermis and inversely correlated with expression of the endogenous angiogenesis inhibitor, IFN-beta. Int J Oncol 14 (3): 401-8, 1999.
- Colonna V, Resta L, Napoli A, et al.: Placental hypoxia and neonatal haemangioma: clinical and histological observations. Br J Dermatol 162 (1): 208-9, 2010.
- de Jong S, Itinteang T, Withers AH, et al.: Does hypoxia play a role in infantile hemangioma? Arch Dermatol Res 308 (4): 219-27, 2016.
- North PE, Waner M, Mizeracki A, et al.: A unique microvascular phenotype shared by juvenile hemangiomas and human placenta. Arch Dermatol 137 (5): 559-70, 2001.
- Dubois J, Patriquin HB, Garel L, et al.: Soft-tissue hemangiomas in infants and children: diagnosis using Doppler sonography. AJR Am J Roentgenol 171 (1): 247-52, 1998.
- Horii KA, Drolet BA, Frieden IJ, et al.: Prospective study of the frequency of hepatic hemangiomas in infants with multiple cutaneous infantile hemangiomas. Pediatr Dermatol 28 (3): 245-53, 2011 May-Jun.
- Ma EH, Robertson SJ, Chow CW, et al.: Infantile Hemangioma with Minimal or Arrested Growth: Further Observations on Clinical and Histopathologic Findings of this Unique but Underrecognized Entity. Pediatr Dermatol 34 (1): 64-71, 2017.
- Valdivielso-Ramos M, Torrelo A, Martin-Santiago A, et al.: Infantile hemangioma with minimal or arrested growth as the skin manifestation of PHACE syndrome. Pediatr Dermatol 35 (5): 622-627, 2018.
- Planas-Ciudad S, Roé Crespo E, Sánchez-Carpintero I, et al.: Infantile hemangiomas with minimal or arrested growth associated with soft tissue hypertrophy: a case series of 10 patients. J Eur Acad Dermatol Venereol 31 (11): 1924-1929, 2017.
- Elluru RG, Friess MR, Richter GT, et al.: Multicenter Evaluation of the Effectiveness of Systemic Propranolol in the Treatment of Airway Hemangiomas. Otolaryngol Head Neck Surg 153 (3): 452-60, 2015.
- McCormick AA, Tarchichi T, Azbell C, et al.: Subglottic hemangioma: Understanding the association with facial segmental hemangioma in a beard distribution. Int J Pediatr Otorhinolaryngol 113: 34-37, 2018.
- Xue L, Sun C, Xu DP, et al.: Clinical Outcomes of Infants With Periorbital Hemangiomas Treated With Oral Propranolol. J Oral Maxillofac Surg 74 (11): 2193-2199, 2016.
- Proisy M, Powell J, McCuaig C, et al.: PHACES Syndrome and Associated Anomalies: Risk Associated With Small and Large Facial Hemangiomas. AJR Am J Roentgenol 217 (2): 507-514, 2021.
- Theiler M, Baselga E, Gerth-Kahlert C, et al.: Infantile hemangiomas with conjunctival involvement: An underreported occurrence. Pediatr Dermatol 34 (6): 681-685, 2017.
- Garzon MC, Epstein LG, Heyer GL, et al.: PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr 178: 24-33.e2, 2016.
- Frieden IJ, Reese V, Cohen D: PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol 132 (3): 307-11, 1996.
- Metry D, Heyer G, Hess C, et al.: Consensus Statement on Diagnostic Criteria for PHACE Syndrome. Pediatrics 124 (5): 1447-56, 2009.
- Metry DW, Haggstrom AN, Drolet BA, et al.: A prospective study of PHACE syndrome in infantile hemangiomas: demographic features, clinical findings, and complications. Am J Med Genet A 140 (9): 975-86, 2006.
- Drolet BA, Dohil M, Golomb MR, et al.: Early stroke and cerebral vasculopathy in children with facial hemangiomas and PHACE association. Pediatrics 117 (3): 959-64, 2006.
- Heyer GL, Dowling MM, Licht DJ, et al.: The cerebral vasculopathy of PHACES syndrome. Stroke 39 (2): 308-16, 2008.
- Haggstrom AN, Garzon MC, Baselga E, et al.: Risk for PHACE syndrome in infants with large facial hemangiomas. Pediatrics 126 (2): e418-26, 2010.
- Poindexter G, Metry DW, Barkovich AJ, et al.: PHACE syndrome with intracerebral hemangiomas, heterotopia, and endocrine dysfunction. Pediatr Neurol 36 (6): 402-6, 2007.
- Brandon K, Burrows P, Hess C, et al.: Arteriovenous malformation: a rare manifestation of PHACE syndrome. Pediatr Dermatol 28 (2): 180-4, 2011 Mar-Apr.
- Chan YC, Eichenfield LF, Malchiodi J, et al.: Small facial haemangioma and supraumbilical raphe--a forme fruste of PHACES syndrome? Br J Dermatol 153 (5): 1053-7, 2005.
- Nabatian AS, Milgraum SS, Hess CP, et al.: PHACE without face? Infantile hemangiomas of the upper body region with minimal or absent facial hemangiomas and associated structural malformations. Pediatr Dermatol 28 (3): 235-41, 2011 May-Jun.
- Antonov NK, Spence-Shishido A, Marathe KS, et al.: Orbital Hemangioma with Intracranial Vascular Anomalies and Hemangiomas: A New Presentation of PHACE Syndrome? Pediatr Dermatol 32 (6): e267-72, 2015 Nov-Dec.
- Burrows PE, Robertson RL, Mulliken JB, et al.: Cerebral vasculopathy and neurologic sequelae in infants with cervicofacial hemangioma: report of eight patients. Radiology 207 (3): 601-7, 1998.
- Hess CP, Fullerton HJ, Metry DW, et al.: Cervical and intracranial arterial anomalies in 70 patients with PHACE syndrome. AJNR Am J Neuroradiol 31 (10): 1980-6, 2010.
- Yu J, Siegel DH, Drolet BA, et al.: Prevalence and Clinical Characteristics of Headaches in PHACE Syndrome. J Child Neurol 31 (4): 468-73, 2016.
- Samuelov L, Kinori M, Mancini AJ, et al.: Ocular Complications in PHACE Syndrome: A True Association or a Coincidence? J Pediatr 204: 214-218.e2, 2019.
- Stefanko NS, Davies OMT, Beato MJ, et al.: Hamartomas and midline anomalies in association with infantile hemangiomas, PHACE, and LUMBAR syndromes. Pediatr Dermatol 37 (1): 78-85, 2020.
- Martin KL, Arvedson JC, Bayer ML, et al.: Risk of dysphagia and speech and language delay in PHACE syndrome. Pediatr Dermatol 32 (1): 64-9, 2015 Jan-Feb.
- Chiu YE, Siegel DH, Drolet BA, et al.: Tooth enamel hypoplasia in PHACE syndrome. Pediatr Dermatol 31 (4): 455-8, 2014 Jul-Aug.
- Duffy KJ, Runge-Samuelson C, Bayer ML, et al.: Association of hearing loss with PHACE syndrome. Arch Dermatol 146 (12): 1391-6, 2010.
- Letertre O, Boccara O, Prey S, et al.: Segmental facial infantile haemangiomas in the era of propranolol: evaluation at 6 years of age. J Eur Acad Dermatol Venereol 36 (4): 610-614, 2022.
- Braun M, Frieden IJ, Siegel DH, et al.: Multicenter Study of Long-Term Outcomes and Quality of Life in PHACE Syndrome after Age 10. J Pediatr 267: 113907, 2024.
- Iacobas I, Burrows PE, Frieden IJ, et al.: LUMBAR: association between cutaneous infantile hemangiomas of the lower body and regional congenital anomalies. J Pediatr 157 (5): 795-801.e1-7, 2010.
- Girard C, Bigorre M, Guillot B, et al.: PELVIS Syndrome. Arch Dermatol 142 (7): 884-8, 2006.
- Stockman A, Boralevi F, Taïeb A, et al.: SACRAL syndrome: spinal dysraphism, anogenital, cutaneous, renal and urologic anomalies, associated with an angioma of lumbosacral localization. Dermatology 214 (1): 40-5, 2007.
- Anwar T, Malm-Buatsi E: Propranolol as an effective therapy for infantile haemangioma of the urinary bladder. BMJ Case Rep 12 (2): , 2019.
- Drolet BA, Chamlin SL, Garzon MC, et al.: Prospective study of spinal anomalies in children with infantile hemangiomas of the lumbosacral skin. J Pediatr 157 (5): 789-94, 2010.
- Sardana K, Gupta R, Garg VK, et al.: A prospective study of cutaneous manifestations of spinal dysraphism from India. Pediatr Dermatol 26 (6): 688-95, 2009 Nov-Dec.
- Rialon KL, Murillo R, Fevurly RD, et al.: Risk factors for mortality in patients with multifocal and diffuse hepatic hemangiomas. J Pediatr Surg 50 (5): 837-41, 2015.
- Hsi Dickie B, Fishman SJ, Azizkhan RG: Hepatic vascular tumors. Semin Pediatr Surg 23 (4): 168-72, 2014.
- Ji Y, Chen S, Yang K, et al.: Screening for infantile hepatic hemangioma in patients with cutaneous infantile hemangioma: A multicenter prospective study. J Am Acad Dermatol 84 (5): 1378-1384, 2021.
- Huang SA, Tu HM, Harney JW, et al.: Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med 343 (3): 185-9, 2000.
- Fernández Faith E, Shah S, Witman PM, et al.: Clinical Features, Prognostic Factors, and Treatment Interventions for Ulceration in Patients With Infantile Hemangioma. JAMA Dermatol 157 (5): 566-572, 2021.
- de Graaf M, Knol MJ, Totté JE, et al.: E-learning enables parents to assess an infantile hemangioma. J Am Acad Dermatol 70 (5): 893-8, 2014.
- Léauté-Labrèze C, Baselga Torres E, Weibel L, et al.: The Infantile Hemangioma Referral Score: A Validated Tool for Physicians. Pediatrics 145 (4): , 2020.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al.: Clinical Practice Guideline for the Management of Infantile Hemangiomas. Pediatrics 143 (1): , 2019.
- Kessels JP, Hamers ET, Ostertag JU: Superficial hemangioma: pulsed dye laser versus wait-and-see. Dermatol Surg 39 (3 Pt 1): 414-21, 2013.
- Trapeznikova TV, Pisklakova TP, Khomchenko VV, et al.: New technology for coagulation of dilated vessels using the combined effects of several modes of generation and wavelengths in one laser pulse for the treatment of pediatric hemangiomas: Open prospective study. Dermatol Ther 33 (3): e13341, 2020.
- He HY, Shi WK, Jiang JC, et al.: An exploration of optimal time and safety of 595-nm pulsed dye laser for the treatment of early superficial infantile hemangioma. Dermatol Ther 35 (5): e15406, 2022.
- Keller RG, Patel KG: Evidence-Based Medicine in the Treatment of Infantile Hemangiomas. Facial Plast Surg Clin North Am 23 (3): 373-92, 2015.
- Sharifpanah F, Saliu F, Bekhite MM, et al.: β-Adrenergic receptor antagonists inhibit vasculogenesis of embryonic stem cells by downregulation of nitric oxide generation and interference with VEGF signalling. Cell Tissue Res 358 (2): 443-52, 2014.
- Ma X, Zhao T, Ouyang T, et al.: Propranolol enhanced adipogenesis instead of induction of apoptosis of hemangiomas stem cells. Int J Clin Exp Pathol 7 (7): 3809-17, 2014.
- Sasaki M, North PE, Elsey J, et al.: Propranolol exhibits activity against hemangiomas independent of beta blockade. NPJ Precis Oncol 3: 27, 2019.
- Overman J, Fontaine F, Wylie-Sears J, et al.: R-propranolol is a small molecule inhibitor of the SOX18 transcription factor in a rare vascular syndrome and hemangioma. Elife 8: , 2019.
- Seebauer CT, Graus MS, Huang L, et al.: Non-beta blocker enantiomers of propranolol and atenolol inhibit vasculogenesis in infantile hemangioma. J Clin Invest 132 (3): , 2022.
- Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, et al.: A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med 372 (8): 735-46, 2015.
- Bauman NM: Propanolol effectively treats significant infantile hemangiomas. J Pediatr 167 (1): 210, 2015.
- Chang L, Ye X, Qiu Y, et al.: Is Propranolol Safe and Effective for Outpatient Use for Infantile Hemangioma? A Prospective Study of 679 Cases From One Center in China. Ann Plast Surg 76 (5): 559-63, 2016.
- Ames JA, Sykes JM: Current trends in medical management of infantile hemangioma. Curr Opin Otolaryngol Head Neck Surg 23 (4): 286-91, 2015.
- Lou Y, Peng WJ, Cao Y, et al.: The effectiveness of propranolol in treating infantile haemangiomas: a meta-analysis including 35 studies. Br J Clin Pharmacol 78 (1): 44-57, 2014.
- Luo Y, Zeng Y, Zhou B, et al.: A retrospective study of propranolol therapy in 635 infants with infantile hemangioma. Pediatr Dermatol 32 (1): 151-2, 2015 Jan-Feb.
- Vivas-Colmenares GV, Bernabeu-Wittel J, Alonso-Arroyo V, et al.: Effectiveness of propranolol in the treatment of infantile hemangioma beyond the proliferation phase. Pediatr Dermatol 32 (3): 348-52, 2015 May-Jun.
- Kridin K, Pam N, Bergman R, et al.: Oral propranolol administration is effective for infantile hemangioma in late infancy: A retrospective cohort study. Dermatol Ther 33 (3): e13331, 2020.
- Liu X, Qu X, Zheng J, et al.: Effectiveness and Safety of Oral Propranolol versus Other Treatments for Infantile Hemangiomas: A Meta-Analysis. PLoS One 10 (9): e0138100, 2015.
- Hardison S, Wan W, Dodson KM: The use of propranolol in the treatment of subglottic hemangiomas: A literature review and meta-analysis. Int J Pediatr Otorhinolaryngol 90: 175-180, 2016.
- Mehta A, Bajaj MS, Pushker N, et al.: To compare intralesional and oral propranolol for treating periorbital and eyelid capillary hemangiomas. Indian J Ophthalmol 67 (12): 1974-1980, 2019.
- Drolet BA, Frommelt PC, Chamlin SL, et al.: Initiation and use of propranolol for infantile hemangioma: report of a consensus conference. Pediatrics 131 (1): 128-40, 2013.
- Solman L, Glover M, Beattie PE, et al.: Oral propranolol in the treatment of proliferating infantile haemangiomas: British Society for Paediatric Dermatology consensus guidelines. Br J Dermatol 179 (3): 582-589, 2018.
- Hoeger PH, Harper JI, Baselga E, et al.: Treatment of infantile haemangiomas: recommendations of a European expert group. Eur J Pediatr 174 (7): 855-65, 2015.
- Raphael MF, Breugem CC, Vlasveld FA, et al.: Is cardiovascular evaluation necessary prior to and during beta-blocker therapy for infantile hemangiomas?: A cohort study. J Am Acad Dermatol 72 (3): 465-72, 2015.
- Streicher JL, Riley EB, Castelo-Soccio LA: Reevaluating the Need for Electrocardiograms Prior to Initiation of Treatment With Propranolol for Infantile Hemangiomas. JAMA Pediatr 170 (9): 906-7, 2016.
- Huang CY, Perman MJ, Yan AC: Regimen for accelerated propranolol initial dosing (RAPID). Pediatr Dermatol 41 (4): 621-627, 2024.
- Püttgen KB, Hansen LM, Lauren C, et al.: Limited utility of repeated vital sign monitoring during initiation of oral propranolol for complicated infantile hemangioma. J Am Acad Dermatol 85 (2): 345-352, 2021.
- Olsen GM, Hansen LM, Stefanko NS, et al.: Evaluating the Safety of Oral Propranolol Therapy in Patients With PHACE Syndrome. JAMA Dermatol 156 (2): 186-190, 2020.
- Prey S, Voisard JJ, Delarue A, et al.: Safety of Propranolol Therapy for Severe Infantile Hemangioma. JAMA 315 (4): 413-5, 2016.
- Morimoto A, Ozeki M, Sasaki S, et al.: Severe hypoglycemia in propranolol treatment for infantile hemangiomas. Pediatr Int 64 (1): e15278, 2022.
- Wedgeworth E, Glover M, Irvine AD, et al.: Propranolol in the treatment of infantile haemangiomas: lessons from the European Propranolol In the Treatment of Complicated Haemangiomas (PITCH) Taskforce survey. Br J Dermatol 174 (3): 594-601, 2016.
- Ji Y, Chen S, Wang Q, et al.: Intolerable side effects during propranolol therapy for infantile hemangioma: frequency, risk factors and management. Sci Rep 8 (1): 4264, 2018.
- Baselga E, Dembowska-Baginska B, Przewratil P, et al.: Efficacy of Propranolol Between 6 and 12 Months of Age in High-Risk Infantile Hemangioma. Pediatrics 142 (3): , 2018.
- Shah SD, Baselga E, McCuaig C, et al.: Rebound Growth of Infantile Hemangiomas After Propranolol Therapy. Pediatrics 137 (4): , 2016.
- Frongia G, Byeon JO, Mehrabi A, et al.: Recurrence rate of infantile hemangioma after oral propranolol therapy. Eur J Pediatr 180 (2): 585-590, 2021.
- O'Brien KF, Shah SD, Pope E, et al.: Late growth of infantile hemangiomas in children >3 years of age: A retrospective study. J Am Acad Dermatol 80 (2): 493-499, 2019.
- Phillips RJ, Crock CM, Penington AJ, et al.: Prolonged tumour growth after treatment of infantile haemangioma with propranolol. Med J Aust 206 (3): 131, 2017.
- Ábarzúa-Araya A, Navarrete-Dechent CP, Heusser F, et al.: Atenolol versus propranolol for the treatment of infantile hemangiomas: a randomized controlled study. J Am Acad Dermatol 70 (6): 1045-9, 2014.
- Bayart CB, Tamburro JE, Vidimos AT, et al.: Atenolol Versus Propranolol for Treatment of Infantile Hemangiomas During the Proliferative Phase: A Retrospective Noninferiority Study. Pediatr Dermatol 34 (4): 413-421, 2017.
- Pope E, Lara-Corrales I, Sibbald C, et al.: Noninferiority and Safety of Nadolol vs Propranolol in Infants With Infantile Hemangioma: A Randomized Clinical Trial. JAMA Pediatr 176 (1): 34-41, 2022.
- Ji Y, Wang Q, Chen S, et al.: Oral atenolol therapy for proliferating infantile hemangioma: A prospective study. Medicine (Baltimore) 95 (24): e3908, 2016.
- McGillis E, Baumann T, LeRoy J: Death Associated With Nadolol for Infantile Hemangioma: A Case for Improving Safety. Pediatrics 145 (1): , 2020.
- Bernabeu-Wittel J, Narváez-Moreno B, de la Torre-García JM, et al.: Oral Nadolol for Children with Infantile Hemangiomas and Sleep Disturbances with Oral Propranolol. Pediatr Dermatol 32 (6): 853-7, 2015 Nov-Dec.
- Chinnadurai S, Fonnesbeck C, Snyder KM, et al.: Pharmacologic Interventions for Infantile Hemangioma: A Meta-analysis. Pediatrics 137 (2): e20153896, 2016.
- Xu DP, Cao RY, Tong S, et al.: Topical timolol maleate for superficial infantile hemangiomas: an observational study. J Oral Maxillofac Surg 73 (6): 1089-94, 2015.
- Tawfik AA, Alsharnoubi J: Topical timolol solution versus laser in treatment of infantile hemangioma: a comparative study. Pediatr Dermatol 32 (3): 369-76, 2015 May-Jun.
- Püttgen K, Lucky A, Adams D, et al.: Topical Timolol Maleate Treatment of Infantile Hemangiomas. Pediatrics 138 (3): , 2016.
- Drolet BA, Boakye-Agyeman F, Harper B, et al.: Systemic timolol exposure following topical application to infantile hemangiomas. J Am Acad Dermatol 82 (3): 733-736, 2020.
- Weibel L, Barysch MJ, Scheer HS, et al.: Topical Timolol for Infantile Hemangiomas: Evidence for Efficacy and Degree of Systemic Absorption. Pediatr Dermatol 33 (2): 184-90, 2016 Mar-Apr.
- Frommelt P, Juern A, Siegel D, et al.: Adverse Events in Young and Preterm Infants Receiving Topical Timolol for Infantile Hemangioma. Pediatr Dermatol 33 (4): 405-14, 2016.
- Xia M, Ding K, Ji Y, et al.: The timing and safety of topical timolol treatment for superficial infantile hemangioma: a retrospective cohort study. Eur J Pediatr 184 (2): 151, 2025.
- Muñoz-Garza FZ, Ríos M, Roé-Crespo E, et al.: Efficacy and Safety of Topical Timolol for the Treatment of Infantile Hemangioma in the Early Proliferative Stage: A Randomized Clinical Trial. JAMA Dermatol 157 (5): 583-587, 2021.
- Aly MM, Hamza AF, Abdel Kader HM, et al.: Therapeutic superiority of combined propranolol with short steroids course over propranolol monotherapy in infantile hemangioma. Eur J Pediatr 174 (11): 1503-9, 2015.
- Li G, Xu DP, Tong S, et al.: Oral Propranolol With Topical Timolol Maleate Therapy for Mixed Infantile Hemangiomas in Oral and Maxillofacial Regions. J Craniofac Surg 27 (1): 56-60, 2016.
- Tong S, Xu DP, Liu ZM, et al.: Evaluation of the efficacy and safety of topical timolol maleate combined with oral propranolol treatment for parotid mixed infantile hemangiomas. Oncol Lett 12 (3): 1806-1810, 2016.
- Ge J, Zheng J, Zhang L, et al.: Oral propranolol combined with topical timolol for compound infantile hemangiomas: a retrospective study. Sci Rep 6: 19765, 2016.
- Frieden IJ, Püttgen KB, Drolet BA, et al.: Management of infantile hemangiomas during the COVID pandemic. Pediatr Dermatol 37 (3): 412-418, 2020.
- Maguiness S, Uihlein LC, Liang MG, et al.: Rapidly involuting congenital hemangioma with fetal involution. Pediatr Dermatol 32 (3): 321-6, 2015 May-Jun.
- Scalise R, Bolton J, Gibbs NF: Rapidly involuting congenital hemangioma (RICH): a brief case report. Dermatol Online J 20 (11): , 2014.
- Kumarasamy MT, Castrisios G, Sharma BK: Rapidly involuting congenital haemangioma in a term neonate. BMJ Case Rep 2014: , 2014.
- Hughes R, McAleer M, Watson R, et al.: Rapidly involuting congenital hemangioma with pustules: two cases. Pediatr Dermatol 31 (3): 398-400, 2014 May-Jun.
- Waelti SL, Rypens F, Damphousse A, et al.: Ultrasound findings in rapidly involuting congenital hemangioma (RICH) - beware of venous ectasia and venous lakes. Pediatr Radiol 48 (4): 586-593, 2018.
- Nasseri E, Piram M, McCuaig CC, et al.: Partially involuting congenital hemangiomas: a report of 8 cases and review of the literature. J Am Acad Dermatol 70 (1): 75-9, 2014.
- Lee PW, Frieden IJ, Streicher JL, et al.: Characteristics of noninvoluting congenital hemangioma: a retrospective review. J Am Acad Dermatol 70 (5): 899-903, 2014.
- Enjolras O, Mulliken JB, Boon LM, et al.: Noninvoluting congenital hemangioma: a rare cutaneous vascular anomaly. Plast Reconstr Surg 107 (7): 1647-54, 2001.
- Vildy S, Macher J, Abasq-Thomas C, et al.: Life-threatening hemorrhaging in neonatal ulcerated congenital hemangioma: two case reports. JAMA Dermatol 151 (4): 422-5, 2015.
- Ayturk UM, Couto JA, Hann S, et al.: Somatic Activating Mutations in GNAQ and GNA11 Are Associated with Congenital Hemangioma. Am J Hum Genet 98 (4): 789-95, 2016.
- WHO Classification of Tumours Editorial Board: WHO Classification of Tumours. Volume 3: Soft Tissue and Bone Tumours. 5th ed., IARC Press, 2020.
- International Society for the Study of Vascular Anomalies: ISSVA Classification of Vascular Anomalies. Milwaukee, Wi: International Society for the Study of Vascular Anomalies, 2018. Available online. Last accessed June 7, 2022.
- Wassef M, Blei F, Adams D, et al.: Vascular Anomalies Classification: Recommendations From the International Society for the Study of Vascular Anomalies. Pediatrics 136 (1): e203-14, 2015.
- Dehner LP, Ishak KG: Vascular tumors of the liver in infants and children. A study of 30 cases and review of the literature. Arch Pathol 92 (2): 101-11, 1971.
- Kulungowski AM, Alomari AI, Chawla A, et al.: Lessons from a liver hemangioma registry: subtype classification. J Pediatr Surg 47 (1): 165-70, 2012.
- Sari N, Yalçin B, Akyüz C, et al.: Infantile hepatic hemangioendothelioma with elevated serum alpha-fetoprotein. Pediatr Hematol Oncol 23 (8): 639-47, 2006.
- Seo IS, Min KW, Mirkin LD: Hepatic hemangioendothelioma of infancy associated with elevated alpha fetoprotein and catecholamine by-products. Pediatr Pathol 8 (6): 625-31, 1988.
- Langham MR, Furman WL, Fernandez-Pineda I: Current Management of Neonatal Liver Tumors. Curr Pediatr Rev 11 (3): 195-204, 2015.
- Berklite L, Malik F, Ranganathan S, et al.: Pediatric hepatic vascular tumors: clinicopathologic characteristics of 33 cases and proposed updates to current classification schemes. Hum Pathol 141: 78-89, 2023.
- Kayaalp C, Sabuncuoglu MZ: Embolization of Liver Hemangiomas. Hepat Mon 15 (8): e30334, 2015.
- Klein M, Chang AK, Vasudevan SA, et al.: Clinically significant ascites as an indication for resection of rapidly involuting congenital hepatic hemangiomas. Pediatr Blood Cancer 65 (8): e27222, 2018.
- Gourgiotis S, Moustafellos P, Zavos A, et al.: Surgical treatment of hepatic haemangiomas: a 15-year experience. ANZ J Surg 76 (9): 792-5, 2006.
- Schmitz R, Heinig J, Klockenbusch W, et al.: Antenatal diagnosis of a giant fetal hepatic hemangioma and treatment with maternal corticosteroid. Ultraschall Med 30 (3): 223-6, 2009.
- Rialon KL, Murillo R, Fevurly RD, et al.: Impact of Screening for Hepatic Hemangiomas in Patients with Multiple Cutaneous Infantile Hemangiomas. Pediatr Dermatol 32 (6): 808-12, 2015 Nov-Dec.
- Yeh I, Bruckner AL, Sanchez R, et al.: Diffuse infantile hepatic hemangiomas: a report of four cases successfully managed with medical therapy. Pediatr Dermatol 28 (3): 267-75, 2011 May-Jun.
- Wasserman JD, Mahant S, Carcao M, et al.: Vincristine for successful treatment of steroid-dependent infantile hemangiomas. Pediatrics 135 (6): e1501-5, 2015.
- Vlahovic A, Simic R, Djokic D, et al.: Diffuse neonatal hemangiomatosis treatment with cyclophosphamide: a case report. J Pediatr Hematol Oncol 31 (11): 858-60, 2009.
- Sundar Alagusundaramoorthy S, Vilchez V, Zanni A, et al.: Role of transplantation in the treatment of benign solid tumors of the liver: a review of the United Network of Organ Sharing data set. JAMA Surg 150 (4): 337-42, 2015.
- Jeng MR, Fuh B, Blatt J, et al.: Malignant transformation of infantile hemangioma to angiosarcoma: response to chemotherapy with bevacizumab. Pediatr Blood Cancer 61 (11): 2115-7, 2014.
- Rutten C, Ackermann O, Lambert V, et al.: Pediatric hepatic hemangiomas: spectrum and prognostic significance of initial ultrasound findings. Pediatr Radiol 53 (12): 2446-2457, 2023.
- Lucas B, Ravishankar S, Pateva I: Pediatric Primary Hepatic Tumors: Diagnostic Considerations. Diagnostics (Basel) 11 (2): , 2021.
- Kou K, Chen YG, Zhou JP, et al.: Hepatic epithelioid hemangioendothelioma: Update on diagnosis and therapy. World J Clin Cases 8 (18): 3978-3987, 2020.
- Perkins P, Weiss SW: Spindle cell hemangioendothelioma. An analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol 20 (10): 1196-204, 1996.
- Fletcher CD, Beham A, Schmid C: Spindle cell haemangioendothelioma: a clinicopathological and immunohistochemical study indicative of a non-neoplastic lesion. Histopathology 18 (4): 291-301, 1991.
- Enjolras O, Mulliken JB, Kozakewich HPW: Vascular tumors and tumor-like lesions. In: Mulliken JB, Burrows PE, Fishman SJ, eds.: Mulliken & Young's Vascular Anomalies: Hemangiomas and Malformations. 2nd ed. Oxford University Press, 2013, pp 259-324.
- Hoeger PH, Colmenero I: Vascular tumours in infants. Part I: benign vascular tumours other than infantile haemangioma. Br J Dermatol 171 (3): 466-73, 2014.
- Pansuriya TC, van Eijk R, d'Adamo P, et al.: Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet 43 (12): 1256-61, 2011.
- Guo R, Gavino AC: Angiolymphoid hyperplasia with eosinophilia. Arch Pathol Lab Med 139 (5): 683-6, 2015.
- O'Connell JX, Nielsen GP, Rosenberg AE: Epithelioid vascular tumors of bone: a review and proposal of a classification scheme. Adv Anat Pathol 8 (2): 74-82, 2001.
- Huang SC, Zhang L, Sung YS, et al.: Frequent FOS Gene Rearrangements in Epithelioid Hemangioma: A Molecular Study of 58 Cases With Morphologic Reappraisal. Am J Surg Pathol 39 (10): 1313-21, 2015.
- Liu KX, Duggan EM, Al-Ibraheemi A, et al.: Characterization of long-term outcomes for pediatric patients with epithelioid hemangioma. Pediatr Blood Cancer 66 (1): e27451, 2019.
- Browning JC, Eldin KW, Kozakewich HP, et al.: Congenital disseminated pyogenic granuloma. Pediatr Dermatol 26 (3): 323-7, 2009 May-Jun.
- Putra J, Rymeski B, Merrow AC, et al.: Four cases of pediatric deep-seated/subcutaneous pyogenic granuloma: Review of literature and differential diagnosis. J Cutan Pathol 44 (6): 516-522, 2017.
- Alomari MH, Kozakewich HPW, Kerr CL, et al.: Congenital Disseminated Pyogenic Granuloma: Characterization of an Aggressive Multisystemic Disorder. J Pediatr 226: 157-166, 2020.
- Groesser L, Peterhof E, Evert M, et al.: BRAF and RAS Mutations in Sporadic and Secondary Pyogenic Granuloma. J Invest Dermatol 136 (2): 481-6, 2016.
- Lee J, Sinno H, Tahiri Y, et al.: Treatment options for cutaneous pyogenic granulomas: a review. J Plast Reconstr Aesthet Surg 64 (9): 1216-20, 2011.
- Patrizi A, Gurioli C, Dika E: Pyogenic granulomas in childhood: New treatment modalities. Dermatol Ther 28 (5): 332, 2015 Sep-Oct.
- Oke I, Alkharashi M, Petersen RA, et al.: Treatment of Ocular Pyogenic Granuloma With Topical Timolol. JAMA Ophthalmol 135 (4): 383-385, 2017.
- Jaiswal H, Patidar N, Shah C, et al.: Topical timolol 0.5% as the primary treatment of ophthalmic pyogenic granuloma: A prospective, single-arm study. Indian J Ophthalmol 69 (5): 1155-1160, 2021.
- Neri I, Baraldi C, Balestri R, et al.: Topical 1% propranolol ointment with occlusion in treatment of pyogenic granulomas: An open-label study in 22 children. Pediatr Dermatol 35 (1): 117-120, 2018.
- Haemel AK, O'Brian AL, Teng JM: Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol 146 (7): 715-8, 2010.
- Pignatti M, Spaggiari A, Sala P, et al.: Laser treatment of angiofibromas in tuberous sclerosis. Minerva Pediatr 66 (6): 585-6, 2014.
- Lee YI, Lee JH, Kim DY, et al.: Comparative Effects of Topical 0.2% Sirolimus for Angiofibromas in Adults and Pediatric Patients with Tuberous Sclerosis Complex. Dermatology 234 (1-2): 13-22, 2018.
- Wataya-Kaneda M, Ohno Y, Fujita Y, et al.: Sirolimus Gel Treatment vs Placebo for Facial Angiofibromas in Patients With Tuberous Sclerosis Complex: A Randomized Clinical Trial. JAMA Dermatol 154 (7): 781-788, 2018.
- Koenig MK, Bell CS, Hebert AA, et al.: Efficacy and Safety of Topical Rapamycin in Patients With Facial Angiofibromas Secondary to Tuberous Sclerosis Complex: The TREATMENT Randomized Clinical Trial. JAMA Dermatol 154 (7): 773-780, 2018.
- Coutinho-Camillo CM, Brentani MM, Nagai MA: Genetic alterations in juvenile nasopharyngeal angiofibromas. Head Neck 30 (3): 390-400, 2008.
- Riggs S, Orlandi RR: Juvenile nasopharyngeal angiofibroma recurrence associated with exogenous testosterone therapy. Head Neck 32 (6): 812-5, 2010.
- Liu Z, Wang J, Wang H, et al.: Hormonal receptors and vascular endothelial growth factor in juvenile nasopharyngeal angiofibroma: immunohistochemical and tissue microarray analysis. Acta Otolaryngol 135 (1): 51-7, 2015.
- Sakthivel P, Kumar R, Arunraj ST, et al.: 68 Ga DOTANOC PET/CT Scan in Primary Juvenile Nasopharyngeal Angiofibroma - A Pilot Study. Laryngoscope 131 (7): 1509-1515, 2021.
- Szymańska A, Szymański M, Czekajska-Chehab E, et al.: Two types of lateral extension in juvenile nasopharyngeal angiofibroma: diagnostic and therapeutic management. Eur Arch Otorhinolaryngol 272 (1): 159-66, 2015.
- Samanta D: Topical mTOR (mechanistic target of rapamycin) inhibitor therapy in facial angiofibroma. Indian J Dermatol Venereol Leprol 81 (5): 540-1, 2015 Sep-Oct.
- Krakowski AC, Nguyen TA: Inhibition of Angiofibromas in a Tuberous Sclerosis Patient Using Topical Timolol 0.5% Gel. Pediatrics 136 (3): e709-13, 2015.
- Mallick S, Benson R, Bhasker S, et al.: Long-term treatment outcomes of juvenile nasopharyngeal angiofibroma treated with radiotherapy. Acta Otorhinolaryngol Ital 35 (2): 75-9, 2015.
- Peters T, Traboulsi D, Tibbles LA, et al.: Sirolimus: a therapeutic advance for dermatologic disease. Skin Therapy Lett 19 (4): 1-4, 2014 Jul-Aug.
- Fernández KS, de Alarcon A, Adams DM, et al.: Sirolimus for the treatment of juvenile nasopharyngeal angiofibroma. Pediatr Blood Cancer 67 (4): e28162, 2020.
Intermediate Tumors (Locally Aggressive)
Kaposiform Hemangioendothelioma and Tufted Angioma
Kaposiform hemangioendothelioma (KHE) and tufted angioma are rare vascular tumors that typically occur during infancy or early childhood but have been reported in adults. Both tumors are thought to be a spectrum of the same disease, because both can be locally aggressive and cause Kasabach-Merritt phenomenon, a serious life-threatening coagulopathy characterized by profound thrombocytopenia and hypofibrinogenemia. They are discussed here as a single entity, kaposiform hemangioendothelioma.
Incidence
The exact incidence of kaposiform hemangioendothelioma is unknown but is estimated to be 0.07 cases per 100,000 children per year.[1,2,3] This lesion affects both sexes equally, with most developing in the neonatal period, one-half presenting at birth, and others presenting during childhood or adulthood.[4]
Clinical presentation
Kaposiform hemangioendothelioma most frequently involves the extremities and less frequently involves the trunk and head and neck area.[3] Most lesions involve the skin (see Figure 8). Deeper lesions (retroperitoneum, thoracic cavity, and muscle) can appear as a bluish-purpuric hue on the skin, whereas superficial lesions can be firm, purpuric or ecchymotic, and painful. Primary bone lesions may cause pain or other nonspecific findings, even without an obvious mass on physical examination.[5][Level of evidence C2] Lesions are usually unifocal and growth is expansive and contiguous. Local lymph nodes may be involved, but there are no reports of distant metastasis. Rare multifocal presentations have been reported, mostly in the bone.[1,2,3]
Figure 8. Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon. The lesion is indurated, firm, and warm with petechiae and purpura. Credit: Denise Adams, M.D.
Fifty to seventy percent of patients with kaposiform hemangioendothelioma develop Kasabach-Merritt phenomenon (KMP), which is a life-threatening complication. The risk of developing Kasabach-Merritt phenomenon is highest in patients with congenital lesions, lesions larger than 6 to 8 cm, deeper lesions, and when kaposiform hemangioendothelioma arises in the retroperitoneum or mediastinum.[3,6,7] This condition is characterized by profound thrombocytopenia (range, 3,000/µL–60,000/µL) and hypofibrinogenemia (<1 g/L). D-dimer and fibrin degradation products are elevated. Severe anemia can occur secondary to tumor sequestration. Severe hemorrhage is rare; however, trauma (biopsy, surgical procedure), ulceration, infection, or delay in initiating treatment may induce progression to disseminated intravascular coagulation, serious bleeding, and even death. Aggressive replacement of blood products, especially platelets, can increase the size of the lesion, causing significant pain and should only be considered with active bleeding and under the direction of a vascular anomalies specialist.[3] The mortality rate is unclear but it has been reported to be as high as 30%.[3,6]
Histopathology
Kaposiform hemangioendothelioma is characterized by sheets of spindle cells with an infiltrative pattern in the dermis, subcutaneous fat, and muscle. There are often areas of fibrosis, with dilated thin-walled vessels infiltrated around the areas of spindle cells. Mixed within these areas are nests of rounded epithelioid cells of vascular origin and aggregates of capillaries with round or irregularly shaped lumens containing platelet-rich fibrin thrombi. There are usually abnormal lymphatic spaces, either within or at the periphery of the lesion. The rate of mitosis is usually low but can be variable. Tufted angioma is characterized by multiple, discrete lobules of tightly packed capillaries (tufts) scattered in the dermis and sometimes in the subcutis, a so-called cannonball pattern.[8] Mitoses are rare.
The pathogenesis is poorly understood. There is some evidence that kaposiform hemangioendothelioma may be derived from lymphatic endothelium, as the spindle cell expresses the vascular markers CD31 and CD34, the vascular endothelial growth factor receptor-3 (VEGFR-3) (a receptor required for lymphangiogenesis), and the lymphatic markers D2-40 and PROX1.[8,9,10] There is no evidence of association with human herpesvirus 8 infection as is present in Kaposi sarcoma.[10]
Genomic data are limited. There have been reports of a small number of patients with GNA14 variants but not in all cases.[11,12]
High serum levels of angiopoietin-2 (Ang-2) have been found in high-risk patients with kaposiform hemangioendothelioma and kaposiform lymphangiomatosis. The Ang-2 levels have also been noted to decrease in response to therapy with sirolimus, which raises the possibility of an effect on the endothelial cells of the kaposiform hemangioendothelioma tumor.[13] Ang-2 is produced and stored in the endothelial cells and acts as a TEK tyrosine kinase antagonist. Ang-2 can promote neovascularization in conjunction with VEGF, and in humans, Ang-2 is greatly increased in vascular remodeling that occurs with sepsis, inflammation, and lymphangiogenesis.[14] These levels have been used for the diagnosis of vascular tumors and assessment of response to therapy.
Diagnostic evaluation
The diagnosis is based on the combination of clinical, histological, and imaging features. Laboratory evaluation is essential for the diagnosis of Kasabach-Merritt phenomenon. Whenever possible, histological confirmation should be obtained, because prolonged therapy is often needed. However, if clinical and imaging findings are highly suggestive of the diagnosis, deferring biopsy may be an option, but this decision should be reached via an interdisciplinary discussion and approach.
Magnetic resonance imaging (MRI) is the preferred imaging modality, especially for kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon and large lesions. T1-weighted sequences typically show a poorly circumscribed soft tissue mass with soft tissue and dermal thickening and diffuse enhancement with gadolinium. T2-weighted sequences show a diffuse increased signal, with stranding in the subcutaneous fat. Gradient sequences show mildly dilated vessels in and around the soft-tissue mass.[3]
For small and superficial lesions, ultrasonography can be useful for diagnosis and can distinguish tufted angioma from kaposiform hemangioendothelioma. Tufted angiomas are more superficial, with well-defined borders and are hyperechoic. Kaposiform hemangioendothelioma has a more infiltrative pattern, with ill-defined borders and mixed echogenicity. Kaposiform hemangioendotheliomas also have an increased vascular density than do tufted angiomas.[15]
Treatment of kaposiform hemangioendothelioma and tufted angioma
Treatment of uncomplicated kaposiform hemangioendothelioma and tufted angioma
There is no evidence-based standard of care for kaposiform hemangioendotheliomas and tufted angiomas. Treatment varies according to size, location, presence of symptoms, and severity of coagulopathy.
Treatment options for uncomplicated kaposiform hemangioendotheliomas and tufted angiomas include the following:
- Observation.
- Surgical excision.
- Pulse-dye laser.
- Topical agents.
- Propranolol.
- Sirolimus with or without steroid therapy.
Observation is an option for patients with low-risk tumors (i.e., no Kasabach-Merritt phenomenon, small tumor size, asymptomatic). Spontaneous regression and/or stability has been noted.[16]
Kaposiform hemangioendotheliomas and tufted angiomas that are uncomplicated and localized can be treated with surgical excision, pulse-dye laser, or topical agents (steroids, sirolimus, or tacrolimus).[16,17,18]
Propranolol therapy has been reported as a treatment option for patients with kaposiform hemangioendotheliomas based on positive results of propranolol use for other more benign vascular tumors. Results have been mixed, with a report of improved effectiveness using higher doses of propranolol.[19,20] Preliminary results indicate that propranolol should be reserved for patients with kaposiform hemangioendotheliomas without Kasabach-Merritt phenomenon and with smaller, less complicated lesions.
Treatment of complicated kaposiform hemangioendothelioma and tufted angioma
Patients who have Kasabach-Merritt phenomenon and/or functional compromise and are symptomatic need aggressive therapy. An American and Canadian multidisciplinary expert panel published guidelines for the management of complicated kaposiform hemangioendotheliomas.[21] A number of treatment therapies have been reported but none have been uniformly effective.[22,23]
Treatment options for complicated kaposiform hemangioendotheliomas and Kasabach-Merritt phenomenon include the following:
- Vincristine with or without steroid therapy.
- Sirolimus with or without steroid therapy.
- Surgical excision.
Vincristine with or without steroid therapy
The most common treatment option for complicated kaposiform hemangioendotheliomas with or without Kasabach-Merritt phenomenon has traditionally been steroid therapy with or without vincristine or other agents.[21,22,23,24,25,26] However, many institutions are now using the mTOR inhibitor sirolimus, with or without steroid therapy, as primary treatment for high-risk patients.[27,28,29,30,31] Steroid therapy has not been effective as a single agent for complicated kaposiform hemangioendotheliomas, even at high doses. Patients treated with steroid therapy have a response rate of 10% to 20% and a significant number of side effects.[21]
Vincristine was shown to have a hematologic response and reduction in tumor volume in patients with high-risk kaposiform hemangioendotheliomas.[22] Furthermore, in a retrospective review of 37 children with kaposiform hemangioendotheliomas whose lesions did not respond to steroids, 26 of the lesions achieved complete remission, with platelet counts reaching normal levels within 7.6 (± 5.2) weeks after vincristine treatment.[24][Level of evidence C3] Vincristine monotherapy in other studies has not been shown to be effective.[27,31] Successful management of patients with kaposiform hemangioendotheliomas who were treated with vincristine and ticlopidine has also been reported.[32]
In 2013, consensus guidelines for the management of complicated kaposiform hemangioendotheliomas proposed the use of vincristine with or without steroids as first-line therapy. This recommendation was based on available evidence.[21]
Sirolimus with or without steroid therapy
Secondary to promising case reports, case series, and a prospective clinical trial, sirolimus may be considered an alternative first-line therapy for patients with kaposiform hemangioendotheliomas.[28,29,33] There are limited studies investigating the effect of sirolimus on kaposiform hemangioendotheliomas/tufted angiomas without Kasabach-Merritt phenomenon.
Evidence (sirolimus therapy):
- In a prospective study that assessed the efficacy and safety of sirolimus for the treatment of complicated vascular anomalies, 13 patients with kaposiform hemangioendotheliomas were treated with sirolimus.[31]
- In patients with kaposiform hemangioendotheliomas and Kasabach-Merritt phenomenon, ten of ten patients had partial responses, with normalization of their platelet count and fibrinogen at the end of 6 and 12 courses.
- Of the three patients with kaposiform hemangioendotheliomas without Kasabach-Merritt phenomenon, two patients experienced partial responses by the end of course 12, and the third patient with multifocal bony disease had disease progression.
- Side effects were minimal in this group of young patients, and no patient with a kaposiform hemangioendothelioma required a dose adjustment or was removed from the study because of toxicity of sirolimus.
- A retrospective study of sirolimus therapy in patients who had nearly all received previous other treatments reported a complete response rate of 73%. All patients assessed as having Kasabach-Merritt phenomenon had recovery of platelet counts between 1 day and 3 weeks (mean, 1.3 weeks).[34]
- One death occurred in this study from a respiratory infection in a child with a pleural effusion and multifocal disease involving the thoracic cavity. Biopsy was not routinely used in this study to confirm diagnosis, raising the possibility that this child had an unrecognized complex lymphatic anomaly.
- A multicenter, retrospective cohort study analyzed 52 Chinese patients with progressive kaposiform hemangioendotheliomas. Thirty-seven patients (71%) had Kasabach-Merritt phenomenon. Those without Kasabach-Merritt phenomenon received sirolimus alone, and 21 of the patients with Kasabach-Merritt phenomenon received a combination of sirolimus and prednisone.[30]
- Overall, 96% of patients at 6 months and 98% of patients at 12 months demonstrated improvement in notable symptoms and/or had improved complications.
- A single case report of a child with a kaposiform hemangioendothelioma who developed recurrence of pain and fibrosis years after initial therapy and was treated with sirolimus for 26 months observed the following:[33]
- The patient's contracture and range of motion improved, the lesion shrank, and the child was well 2 years later.
- A prospective randomized study of patients with kaposiform hemangioendothelioma associated with Kasabach-Merritt syndrome compared sirolimus monotherapy with sirolimus in combination with prednisolone. The primary outcome was defined as achievement of a durable platelet response (platelet count >100 × 109 /L) at week 4.[35][Level of evidence A3]
- At week 4, a durable platelet response was achieved in 35 of 37 patients who received sirolimus and prednisolone, compared with 24 of 36 patients who received sirolimus monotherapy (difference, 27.9%; 95% CI, 10.0%–44.7%).
- Compared with the sirolimus monotherapy group, the combination treatment group showed improvements in measures of durable platelet responses at all points during the initial 3-week treatment period. The durable platelet responses included median platelet counts during weeks 1 to 4, an increased number of patients achieving fibrinogen stabilization at week 4, and objective lesion responses at 12 months.
- The frequencies of total and serious adverse events were similar in both groups.
- Patients who received combination therapy had lower total disease sequelae and fewer blood transfusions.
Most high-risk patients (kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon) are treated with sirolimus to achieve serum blood levels of 8 to 15 ng/mL.[30,31,36,37]
Supportive care and close monitoring of infants on sirolimus
A case report described two children with kaposiform hemangioendotheliomas and Kasabach-Merritt syndrome who died of pulmonary infections after treatment with sirolimus.[38] Another child who received sirolimus and prednisolone developed Pneumocystis jirovecii pneumonia.[39]P. jirovecii pneumonia prophylaxis and close monitoring of patients on sirolimus (especially infants) is encouraged.
Surgical excision
Surgical excision may be possible for lesions that did not respond to medical management or are life threatening. Embolization may be performed in conjunction with surgery or medical therapy; usually, it is a temporizing measure.[40]
Long-term outcomes
Even with therapy, these lesions do not fully regress and can recur. Worsened symptomatology (pain, inflammation) can occur with age, especially around the time of puberty.[41]
Long-term effects include chronic pain, lymphedema, heart failure, and orthopedic issues.[40,41] These lesions prove to be a difficult dilemma for the practitioner because they have a varied clinical spectrum and response to therapy.
Treatment options under clinical evaluation for kaposiform hemangioendothelioma
Information about National Cancer Institute (NCI)–supported clinical trials can be found on the NCI website. For information about clinical trials sponsored by other organizations, see the ClinicalTrials.gov website.
References:
- Rodriguez V, Lee A, Witman PM, et al.: Kasabach-merritt phenomenon: case series and retrospective review of the mayo clinic experience. J Pediatr Hematol Oncol 31 (7): 522-6, 2009.
- Ryan C, Price V, John P, et al.: Kasabach-Merritt phenomenon: a single centre experience. Eur J Haematol 84 (2): 97-104, 2010.
- Croteau SE, Liang MG, Kozakewich HP, et al.: Kaposiform hemangioendothelioma: atypical features and risks of Kasabach-Merritt phenomenon in 107 referrals. J Pediatr 162 (1): 142-7, 2013.
- Lee B, Chiu M, Soriano T, et al.: Adult-onset tufted angioma: a case report and review of the literature. Cutis 78 (5): 341-5, 2006.
- Kuo C, Warren M, Malvar J, et al.: Kaposiform hemangioendothelioma of the bone in children and adolescents. Pediatr Blood Cancer 69 (1): e29392, 2022.
- Ji Y, Yang K, Peng S, et al.: Kaposiform haemangioendothelioma: clinical features, complications and risk factors for Kasabach-Merritt phenomenon. Br J Dermatol 179 (2): 457-463, 2018.
- Chen C, Yan H, Yao W, et al.: Analysis of Risk Factors for Kasabach Merritt Phenomenom in Children With Kaposiform Hemangioendothelioma. J Pediatr Surg 60 (2): 161932, 2025.
- Enjolras O, Soupre V, Picard A: Uncommon benign infantile vascular tumors. Adv Dermatol 24: 105-24, 2008.
- Zukerberg LR, Nickoloff BJ, Weiss SW: Kaposiform hemangioendothelioma of infancy and childhood. An aggressive neoplasm associated with Kasabach-Merritt syndrome and lymphangiomatosis. Am J Surg Pathol 17 (4): 321-8, 1993.
- Arai E, Kuramochi A, Tsuchida T, et al.: Usefulness of D2-40 immunohistochemistry for differentiation between kaposiform hemangioendothelioma and tufted angioma. J Cutan Pathol 33 (7): 492-7, 2006.
- Lim YH, Bacchiocchi A, Qiu J, et al.: GNA14 Somatic Mutation Causes Congenital and Sporadic Vascular Tumors by MAPK Activation. Am J Hum Genet 99 (2): 443-50, 2016.
- Lim YH, Fraile C, Antaya RJ, et al.: Tufted angioma with associated Kasabach-Merritt phenomenon caused by somatic mutation in GNA14. Pediatr Dermatol 36 (6): 963-964, 2019.
- Le Cras TD, Mobberley-Schuman PS, Broering M, et al.: Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis 20 (1): 163-173, 2017.
- Saharinen P, Eklund L, Alitalo K: Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov 16 (9): 635-661, 2017.
- Gong X, Ying H, Zhang Z, et al.: Ultrasonography and magnetic resonance imaging features of kaposiform hemangioendothelioma and tufted angioma. J Dermatol 46 (10): 835-842, 2019.
- Osio A, Fraitag S, Hadj-Rabia S, et al.: Clinical spectrum of tufted angiomas in childhood: a report of 13 cases and a review of the literature. Arch Dermatol 146 (7): 758-63, 2010.
- Burleigh A, Kanigsberg N, Lam JM: Topical rapamycin (sirolimus) for the treatment of uncomplicated tufted angiomas in two children and review of the literature. Pediatr Dermatol 35 (5): e286-e290, 2018.
- Zhang X, Yang K, Chen S, et al.: Tacrolimus ointment for the treatment of superficial kaposiform hemangioendothelioma and tufted angioma. J Dermatol 46 (10): 898-901, 2019.
- Filippi L, Tamburini A, Berti E, et al.: Successful Propranolol Treatment of a Kaposiform Hemangioendothelioma Apparently Resistant to Propranolol. Pediatr Blood Cancer 63 (7): 1290-2, 2016.
- Wang Z, Li K, Dong K, et al.: Variable response to propranolol treatment of kaposiform hemangioendothelioma, tufted angioma, and Kasabach-Merritt phenomenon. Pediatr Blood Cancer 61 (8): 1518-9, 2014.
- Drolet BA, Trenor CC, Brandão LR, et al.: Consensus-derived practice standards plan for complicated Kaposiform hemangioendothelioma. J Pediatr 163 (1): 285-91, 2013.
- Haisley-Royster C, Enjolras O, Frieden IJ, et al.: Kasabach-merritt phenomenon: a retrospective study of treatment with vincristine. J Pediatr Hematol Oncol 24 (6): 459-62, 2002 Aug-Sep.
- Hauer J, Graubner U, Konstantopoulos N, et al.: Effective treatment of kaposiform hemangioendotheliomas associated with Kasabach-Merritt phenomenon using four-drug regimen. Pediatr Blood Cancer 49 (6): 852-4, 2007.
- Wang Z, Li K, Yao W, et al.: Steroid-resistant kaposiform hemangioendothelioma: a retrospective study of 37 patients treated with vincristine and long-term follow-up. Pediatr Blood Cancer 62 (4): 577-80, 2015.
- Fernandez-Pineda I, Lopez-Gutierrez JC, Ramirez G, et al.: Vincristine-ticlopidine-aspirin: an effective therapy in children with Kasabach-Merritt phenomenon associated with vascular tumors. Pediatr Hematol Oncol 27 (8): 641-5, 2010.
- Fernandez-Pineda I, Lopez-Gutierrez JC, Chocarro G, et al.: Long-term outcome of vincristine-aspirin-ticlopidine (VAT) therapy for vascular tumors associated with Kasabach-Merritt phenomenon. Pediatr Blood Cancer 60 (9): 1478-81, 2013.
- Kai L, Wang Z, Yao W, et al.: Sirolimus, a promising treatment for refractory Kaposiform hemangioendothelioma. J Cancer Res Clin Oncol 140 (3): 471-6, 2014.
- Hammill AM, Wentzel M, Gupta A, et al.: Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer 57 (6): 1018-24, 2011.
- Blatt J, Stavas J, Moats-Staats B, et al.: Treatment of childhood kaposiform hemangioendothelioma with sirolimus. Pediatr Blood Cancer 55 (7): 1396-8, 2010.
- Ji Y, Chen S, Xiang B, et al.: Sirolimus for the treatment of progressive kaposiform hemangioendothelioma: A multicenter retrospective study. Int J Cancer 141 (4): 848-855, 2017.
- Adams DM, Trenor CC, Hammill AM, et al.: Efficacy and Safety of Sirolimus in the Treatment of Complicated Vascular Anomalies. Pediatrics 137 (2): e20153257, 2016.
- López V, Martí N, Pereda C, et al.: Successful management of Kaposiform hemangioendothelioma with Kasabach-Merritt phenomenon using vincristine and ticlopidine. Pediatr Dermatol 26 (3): 365-6, 2009 May-Jun.
- Oza VS, Mamlouk MD, Hess CP, et al.: Role of Sirolimus in Advanced Kaposiform Hemangioendothelioma. Pediatr Dermatol 33 (2): e88-92, 2016 Mar-Apr.
- Wang Z, Yao W, Sun H, et al.: Sirolimus therapy for kaposiform hemangioendothelioma with long-term follow-up. J Dermatol 46 (11): 956-961, 2019.
- Ji Y, Chen S, Zhou J, et al.: Sirolimus plus prednisolone vs sirolimus monotherapy for kaposiform hemangioendothelioma: a randomized clinical trial. Blood 139 (11): 1619-1630, 2022.
- Zhang G, Chen H, Gao Y, et al.: Sirolimus for treatment of Kaposiform haemangioendothelioma with Kasabach-Merritt phenomenon: a retrospective cohort study. Br J Dermatol 178 (5): 1213-1214, 2018.
- Mariani LG, Schmitt IR, Garcia CD, et al.: Low dose sirolimus treatment for refractory tufted angioma and congenital kaposiform hemangioendothelioma, both with Kasabach-Merritt phenomenon. Pediatr Blood Cancer 66 (8): e27810, 2019.
- Ying H, Qiao C, Yang X, et al.: A Case Report of 2 Sirolimus-Related Deaths Among Infants With Kaposiform Hemangioendotheliomas. Pediatrics 141 (Suppl 5): S425-S429, 2018.
- Russell TB, Rinker EK, Dillingham CS, et al.: Pneumocystis Jirovecii Pneumonia During Sirolimus Therapy for Kaposiform Hemangioendothelioma. Pediatrics 141 (Suppl 5): S421-S424, 2018.
- Ji Y, Chen S, Yang K, et al.: Kaposiform hemangioendothelioma: current knowledge and future perspectives. Orphanet J Rare Dis 15 (1): 39, 2020.
- Schaefer BA, Wang D, Merrow AC, et al.: Long-term outcome for kaposiform hemangioendothelioma: A report of two cases. Pediatr Blood Cancer 64 (2): 284-286, 2017.


This information does not replace the advice of a doctor. Ignite Healthwise, LLC disclaims any warranty or liability for your use of this information. Your use of this information means that you agree to the Terms of Use and Privacy Policy. Learn how we develop our content.
Healthwise, Healthwise for every health decision, and the Healthwise logo are trademarks of Ignite Healthwise, LLC.